
Abstract
The present study aimed to determine the protective effects and the underlying mechanisms of unfractionated heparin on lipopolysaccharide (LPS)-induced endotoxemia and lung injury in rats. Rats were injected intravenously with LPS at 6 mg/kg. We examined the therapeutic effects of unfractionated heparin (100 or 300 U/kg) on LPS-induced endotoxemia by dosing intravenously simultaneously after LPS challenge. The animal lung edema degree was evaluated by wet/dry weight ratio. The levels of inflammatory mediators including interleukin-1β (IL-1β) and interleukin-6 (IL-6) were assayed by enzyme-linked immunosorbent assay and quantitative real-time RT-PCR. The activation of nuclear factor-κB (NF-κB) was evaluated by Western blotting. The investigations revealed that treatment with unfractionated heparin can attenuate inflammatory responses in a rat model of LPS-induced acute lung injury, and the effect was much better in 300 U/kg group. The mechanisms by which unfractionated heparin exerts its anti-inflammatory effect are correlated with inhibition of IL-1β and IL-6 production via inactivation of NF-κB.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are associated with high mortality rates despite therapeutic advances [1]. Sepsis is the main cause of ALI or ARDS: 18–40 % of patients with sepsis develop ALI or ARDS [2–4]. The pathogenesis of ALI and ARDS is similar to that of sepsis, as these disease states involve uncontrolled host defense responses that lead to inflammation, endothelial injury, disturbed coagulation, decreased fibrinolysis, and fibro-proliferation [5–8]. The inflammatory process played a key role in the development of ALI, which entangled with coagulation disorder. Recent studies suggest that the use of anticoagulants, such as tissue factor pathway inhibitor, antithrombin, thrombomodulin, activated protein C, and fibrinolytics (plasminogen activators and particularly tissue plasminogen activator), may be useful in the treatment of ALI and ARDS [9–12]. These agents also possess anti-inflammatory and fibrinolytic activities of various capacities. Heparin, as a traditional anticoagulant drug, exerts a broad range of cytoprotective actions, including suppression of inflammation [13], prevention of apoptosis [14], and modulation of vascular adherence [15]. Thus, it may represent a novel strategy for sepsis and ALI treatment.
Nuclear factor-κB (NF-κB) is a universal transcription factor that plays a crucial role in regulating the transcription of over 200 genes, many of which play important roles in the development of septic shock [16]. NF-κB is a collection of dimers composed of various combinations of the members of the NF-κB/Rel family. Heterodimers composed of p65/p50 are the most abundant form of NF-κB in most cell types. In physiologibal conditions, NF-κB exists in the cytosol in an inactive state bound to its physiologic inhibitor-κB (I-κB). Its activation of NF-κB was found to parallel and depend on induced degradation of I-κB, which depends on stimulus-induced phosphorylation of I-κB. Once phosphorylated, I-κB undergoes degradation and NF-κB translocates to the nucleus where it regulates the expression of a variety of genes involved in the inflammatory process [17]. Bacteria and bacterial components such as lipopolysaccharide (LPS) can activate NF-κB through multiple signaling pathways, leading to expression of proinflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, IL-8, which activate NF-κB and its upstream signaling molecules, extending NF-κB-mediated inflammatory response in return [18].
Thus, in the current study, we hypothesized that UFH reduces inflammatory responses in a rat model of LPS-induced ALI, and NF-κB pathway plays a central role.
MATERIALS AND METHODS
Animal Groups
Thirty-two male Wistar rats weighing 250–300 g were provided by the Experimental Animal Center, China Medical University. Rats were housed in a room with a 12-h light/dark cycle for at least 1 week to acclimate to the surroundings, with free access to water and food. All experimental procedures were performed under approval from the Animal Care and Use Committee of China Medical University.
Experimental Protocol
All rats were anesthetized with sodium pentobarbital (40 mg/kg, intraperitoneally) and systemic inflammation was induced by administering a bolus injection of LPS (Escherichia coli serotype 055:B5; Sigma, St. Louis, MO) via the caudal vein at a dose of 6 mg/kg. The 32 rats were randomly and equally divided into four groups (n = 8): normal control group, intravenous administration of the same volume of normal saline; ALI group, animals were given saline alone immediately after the injection of LPS; low-dose UFH group, intravenous administration of 100 U/kg of UFH (Shanghai No. 1 Biochemistry and Pharmaceutical Co., China) was given immediately after LPS injection; high-dose UFH group, intravenous administration of 300 U/kg of UFH was given immediately after LPS injection. UFH is extracted from porcine intestinal mucosa or bovine lung with the mean molecular weight size 12,000 Da. UFH used in this experiment is daily used in clinical studies. Blood samples were obtained under anesthesia from the inferior vena cava 6 h after the LPS injection. EDTA-anticoagulated plasma samples obtained by whole blood centrifugation were stored at −80 °C until assay. The rats were killed by exsanguination from the abdominal aorta 6 h after the injection of reagents. Four rats in each group were used to measure lung wet-to-dry (W/D) weight ratio and pulmonary vascular permeability, whereas the lung tissues of the other four rats were divided into two pieces, left lung immersed in 10 % formalin solution for lung histopathological examination and right lung frozen in liquid nitrogen for quantitative real-time RT-PCR or Western blot analysis.
Plasma Cytokine Levels
Plasma levels of IL-1β and IL-6 were determined by using sandwich enzyme-linked immunosorbent assay (ELISA) kits from R&D Systems (Minneapolis, MN) according to the manufacturer’s instruction.
Myeloperoxidase Activity in Lung
Pulmonary leukocyte infiltration was quantified by myeloperoxidase (MPO) activity as previously described [19] using a commercial kit (Jiancheng Bioengineering Institute, Nanjing, China). Approximately 300 mg samples of lung tissue were homogenized and fluidized in extraction buffer. Five milliliters of homogenate was centrifuged at 20,000 rpm for 15 min at 4 °C, and MPO was extracted from the pellet. MPO activity was expressed in units per gram tissue.
Lung W/D Weight Ratio
The lungs were dissected free of nonpulmonary tissue and cleansed of blood stains with absorbent paper, weighed wet, torrefied in an oven at 80 °C for 72 h, and weighed dry. The ratio of the wet lung to the dry lung was calculated to assess tissue edema.
Pulmonary Microvascular Permeability
Microvascular endothelial permeability was assessed by measuring the Evans blue dyes (EBD) leakage index as a marker. EBD (25 mg/kg) was injected intravenously 30 min before the animals were killed. The organ vasculature was flushed free of blood by gentle infusion of 10 ml of pre-warmed PBS through the left ventricle. Then the lung was dissected and immersed in formamide 1 ml/100 mg for 7 days. After centrifugation, the supernant absorbances at 620 and 740 nm were recorded. Tissue haem pigment contamination was corrected using A 740 readings. Tissue EBD content (in milligrams per gram of dry tissue) was calculated by comparing the tissue supernatant A 620 readings with an EBD standard curve.
Histologic Examination
Fresh lung sections were fixed in 4 % paraformaldehyde and embedded in paraffin; 5 μm-thick sections were stained with hematoxylin and eosin (H&E). A pathologist who was blinded to the experimental groups analyzed the lung injury according to the technique of Smith [20]. Briefly, 24 areas in the lung parenchyma in each animal were graded on a scale of 0–4 (0, absent, appears normal; 1, light; 2, moderate; 3, strong; and 4, intense) for congestion, edema, infiltration of inflammatory cells, and hemorrhage.
Quantitative Real-time RT-PCR
Total RNA was isolated from blood cells using Trizol reagent kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The OD260 and OD260/280 values were measured with spectrophotometer to determine the RNA concentrations. Reverse transcription was performed at 40 °C for 45 min and followed by incubation at 95 °C for 5 min. Quantitative real-time RT-PCR was performed using SYBR Green (Tiangen Biotechnology CO., Ltd., Beijing, China) on an Exicycler™ 96 real-time quantitative thermal block. The PCR primer sequences were designed according to the gene sequences reported in GenBank and were chemically synthesized: IL-1β (forward: 5′-CTT AAA GCC CGC CTG ACA GA-3′; reverse: 5′-TCA GAA TGT GGG AGC GAA TG-3′); IL-6 (forward: 5′-AGG GCT CTT CGG CAA ATG TA-3′; reverse: 5′-GAA GGA ATG CCC ATT AAC AAC AA-3′); β-actin (forward: 5′-GTC CAC CTT CCA GCA GAT GTG-3′; reverse: 5′-GCA TTT GCG GTG GAC GAT-3′). The housekeeping gene β-actin was used for normalization. The ratios of the emissions incorporated into the PCR products of the tested gene to the β-actin products were calculated to evaluate relative changes in the mRNA expression levels of the tested genes.
Preparation of Nuclear Extracts and Cytosolic Extracts
The fresh lung tissue was homogenized in buffer A (10 mmol/l HEPES-NaOH at pH 7.9, 1 mmol/l EDTA, 10 mmol/l KCl, 1.5 mmol/l MgCl2, 1 mmol/l PMSF, 1 mmol/l DTT, and 1 μg/ml leupeptin), and homogenates were incubated on ice for 15 min with gentle agitation. After lysis, the pellet was centrifuged at 14,000 rpm for 10 min. The supernatant was collected and the pellet resuspended in 20 μl of cold lysis buffer A and 0.1 % Nonidet P-40 and incubated on ice for 10 min. The sample was centrifuged at 14,000 rpm for 10 min, supernatant discarded, and nuclear pellet resuspended in 15 μl of cold buffer B (20 mmol/l HEPES-NaOH at pH 7.9, 0.2 mmol/l EDTA, 420 mmol/l NaCl, 1.5 mmol/l MgCl2, 25 % glycerin 0.5 mmol/l PMSF, 0.5 mmol/l DTT, and 1 μg/ml leupeptin). Following centrifugation at 14,000 rpm for 10 min, the supernatant (nuclear extract) was harvested and stored at −70 °C. For preparation of cytosolic extracts, the supernatant was centrifuged at 14,000 rpm for 10 min and protein concentrations were determined by the Bradford assay (Sigma-Aldrich) using bovine serum albumin as standard.
Western Blot Analysis
Equal amounts of protein were loaded per well on a 10 % sodium dodecyl sulfate polyacrylamide gel and then transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5 % nonfat dry milk for 2 h. For cytosolic extracts, membranes were incubated overnight at 4 °C in primary antibody solution of I-κBα (1:500), p-I-κBα (1:500; all from Santa Cruz Biotechnology). For nuclear extracts, membranes were incubated overnight at 4 °C in primary antibody solution of NF-κB p65 (1:1,000 dilution). Membranes were washed three times for 5 min each with TTBS and then incubated with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad Laboratories, Berkeley, CA) for 1 h. Bound antibody was detected by chemiluminescence (ECL Plus kit; Amersham) and high-performance chemiluminescence film. The density of the bands was quantified using Image-J software. The β-actin Western blot was performed as an internal control of protein loading.
Statistical Analysis
Data analysis was performed using SPSS 13.0 software. The results were reported as mean ± SD and repeated at least three times. All results were analyzed by one-way ANOVA with multiple comparisons, and whenever appropriate, analyzed by Newman–Keuls test. The p values lower than 0.05 were considered statistically significant.
RESULTS
Effects of UFH on Lung Pathological Changes of ALI Rats
To evaluate the effects of unfractionated heparin on ALI, we primarily determined the histological changes after unfractionated heparin treatment in LPS exposed rats. Light microscopic findings in the control group included no inflammation change and the thin layer of connective tissue in the lung (Fig. 1a). The lungs in LPS group were significantly damaged; the alveolar wall was thickened, infiltrated by inflammatory cells, and focally hemorrhagic, associated with edematous fluid in the alveolar space (Fig. 1b). UFH treatment attenuated the changes in lung architecture and inflammatory cell infiltration in comparison with the ALI group (Fig. 1c, d). Histological scores were significantly lower in the UFH group than in the LPS group (Fig. 1e).

Effect of UFH on lung histopathological changes of LPS-induced ALI rats (H&E staining; magnification, ×200). Rats were given an intravenous injection of LPS via the caudal vein at a dose of 6 mg/kg or equal amount of normal saline. Some LPS-exposed rats were given UFH (100 or 300 U/kg) immediately after LPS injection. Lungs (n = 4) from each experimental group were processed for histological analysis at 6 h after LPS challenge. a Control group. b ALI group. c UFH 100 U/kg group. d UFH 300 U/kg group. e Histological scores were significantly higher in the ALI group than in the normal control group, heparin treatment decreased the extent of congestion, edema, and inflammation. **p < 0.01 compared with the control group; #p < 0.05; ##p < 0.01 compared with the LPS-treated group.
Effects of UFH on the Lung Edema
To assess tissue water accumulation, tissue W/D weight ratio was measured as an indicator of organ injury. As shown in Fig. 2, the lung W/D weight ratios were obviously increased after LPS administration compared with control group. UFH treatment could reduce the lung W/D weight ratio.

Effects of UFH on lung W/D weight ratios. The lung W/D weight ratios were determined at 6 h after LPS given. The values are presented as means ± SD (n = 4). **p < 0.01 compared with the control group; #P < 0.05; ##P < 0.01 compared with the LPS-treated group.
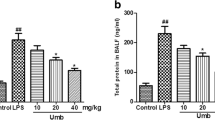
Effect of UFH on Pulmonary Microvascular Permeability
To assess microvascular permeability, we detected the EBD leakage index. Compared with the normal control group, the rats in the ALI group exhibited a significantly increased EBD leakage index in the lungs. The rats in the UFH treatment groups showed a significant reduction in the EBD leakage index (Fig. 3).

Effect of UFH on pulmonary microvascular permeability. Rats were given UFH (100 or 300 U/kg) immediately after LPS injection. EBD content was determined at 6 h after LPS given. The values are presented as means ± SD (n = 4). **p < 0.01 compared with the control group. #p < 0.05; ##p < 0.01 compared with the LPS-treated group.
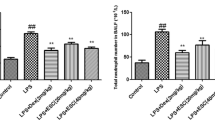
Effect of UFH on Lung Neutrophil Infiltration
To evaluate lung neutrophil infiltration, MPO activity was measured. LPS-treated rats had a significant increase in MPO activity compared with control group. However, UFH-treated rats had significantly lower MPO activity (Fig. 4).

Effect of UFH on lung neutrophil infiltration. Rats were given UFH (100 or 300 U/kg) immediately after LPS injection. MPO activity was determined at 6 h after LPS given. The values are presented as means ± SD (n = 4). **p < 0.01 compared with the control group; #p < 0.05; ##p < 0.01 compared with the LPS-treated group.
Effect of UFH on the Expression of Cytokine in Blood
The concentrations of IL-1β and IL-6 in the blood were thought to play crucial roles in the development of ALI. We enhanced expressions of IL-1β and IL-6 in the blood in the ALI group, and this was prevented by treatment with UFH (Figs. 5a, b and 6a, b)

Effect of UFH on the serum concentrations of IL-1β and IL-6 following LPS injection. IL-1β (a) and IL-6 (b) were determined by means of ELISA at 6 h after LPS given, respectively. The values are presented as means ± SD (n = 4). **p < 0.01 compared with the control group; #p < 0.05; ##p < 0.01 compared with the LPS-treated group.

Effect of UFH on gene expressions of IL-1β and IL-6 in blood cells following LPS injection. Expressions of IL-1β (a) and IL-6 (b) mRNA were analyzed by qRT-PCR (described in “Materials and Methods”) at 6 h after LPS given, respectively. The y-axis represents increase of inflammatory mediators compared with corresponding controls in fold(s). The values are presented as means ± SD (n = 4). **p < 0.01 compared with the control group; #p < 0.05; ##p < 0.01 compared with the LPS-treated group.
Effect of UFH on NF-κB Activation and I-κBα Phosphorylation and Degradation in ALI Rats Induced by LPS
To investigate the anti-inflammatory mechanisms of UFH, we examined the effect of UFH on the nuclear translocation of activated NF-κB p65 subunit in a rat ALI model. As shown in Fig. 7, I-κBα degradation was significantly increased in LPS group, and this increased degradation was blocked by treatment with UFH, which was related to IκBα phosphorylation. LPS-treated rats also showed increase in nuclear extracts of p65 subunit of NF-κB. UFH treatment retained the NF-κB in the cytoplasm even when the amount of LPS used mostly relocated the p65 subunit to the nucleus, especially in UFH 300 U/kg group. Taken together, these results demonstrate that UFH likely exerts an anti-inflammatory effect in part through interfering with the NF-κB-mediated signaling.

Effect of UFH on NF-κB activation (a) and Iκ-Bα phosphorylation and degradation (b) in the lungs of LPS induced ALI rats. The values are presented as means ± SD (n = 4). *p < 0.05; **p < 0.01 compared with the control group; #p < 0.05; ##p < 0.01 compared with the LPS-treated group.
DISCUSSION
Severe sepsis is a common disease and is often complicated with ALI. As such, we selected an LPS-induced ALI animal model to evaluate inflammatory pathways following the administration of heparin. In severe sepsis or septic shock, systemic inflammation response syndrome and coagulation activation synergistically contribute to the subsequent multiple organ dysfunction syndrome [21]. The lung can be injured and its function altered by the coexistence of a hypercoagulable state with an inflammatory reaction in the early stage. These two states interact, potentially aggravating the condition of the patient [22]. Evidences from several clinical studies indicated that proinflammatory cytokines, notably TNF-a, IL-1, and IL-6, participate in the early development of inflammation and lead to pro-coagulant activity; they have been shown to play a crucial role in ALI and ARDS [23]. Heparin is a naturally occurring glycosaminoglycan which has been used as an anticoagulant more than half a century. Recently, a number of clinical and experimental studies have suggested that, aside from their anticoagulant capacity, heparins modulate a wide array of inflammatory responses. Heparin may play a role in inhibiting the production and release of pro-inflammatory factors, including IL-1β, IL-6, and TNF-α [13, 24]. Heparin may inhibit the activation of NF-κB, the pro-inflammatory responses after ischaemia/reperfusion, and attenuate endothelial dysfunction by enhancing nitric oxide and prostacyclin [13]. Similarly, heparin improves endothelial function and vascular reactivity during hyperdynamic sepsis [25]. In the present study, we found that treatment with UFH attenuated the LPS-induced histological alterations in the lung. In addition, UFH had a promising anti-inflammatory activity by showing its abilities to significantly protect rats against LPS-induced ALI. In this study, we found that levels of the cytokines IL-1β and IL-6 at 6 h in blood were dramatically increased after LPS administration. However, treatment of UFH (100 or 300 U/kg) significantly lowered LPS-induced pro-inflammatory cytokines production. Meanwhile, UFH decreased neutrophil infiltration in the lung induced by LPS. This reduction in pro-inflammatory cytokine secretion following LPS activation may be explained at least in part by our recent observation that in vitro, heparin causes a dose-dependent reduction in nuclear translocation of the transcription factor NF-κB [26]. That is, UFH exerted anti-inflammatory effect in vitro and in rats, so it maybe play a similar effect in clinical.
Our study also examined the anti-inflammatory effect of UFH on the pulmonary microvascular permeability. Impaired microvascular endothelial barrier functions in sepsis significantly contribute to organ dysfunction and death [27]. UFH significantly reduced human pulmonary microvascular endothelial cell permeability and decreased the formation of intracellular gaps induced by LPS, by which UFH enhances endothelial barrier function and exerts its protective effect in vitro [28]. In this study, lung W/D weight ratios were measured for evaluating pulmonary edema. The results indicated that the lung W/D weight ratios were the highest in the ALI group and UFH treatment decreased lung edema. The EBD leakage index in the UFH treatment groups was significantly reduced than in the ALI group. Our findings indicate that, at the level of the microvascular endothelium, high-dose UFH appears to exert a greater effect in the expression of an anti-inflammatory phenotype. These in vivo results appear to correlate well with our previous study outcomes in vitro [28].
Among several transcription factors involved in the pathogenesis of sepsis, NF-κB acts as a critical step through the regulation of genes encoding proinflammatory cytokines, such as TNF-α, IL-6, chemokines, adhesion molecules, and other inducible enzymes [18]. To explore the underlying mechanisms by which UFH achieves its beneficial effects, we assessed its influence on NF-κB activation which plays an important role in the pathogenesis of lung diseases. This study demonstrated that NF-κB was activated in LPS-induced lung injury and more importantly, UFH was confirmed not only to inhibit IL-1β and IL-6 production, but also the translocation of NF-κB into the nucleus of the lung by inhibiting the degradation of IκBα. Our previous reports demonstrate that UFH inhibits LPS-induced inflammatory response through blocking p38 MAPK and NF-κB activation on endothelial cell [26]. The anti-inflammatory properties of UFH on endothelial cells were thus supported in vivo.
In summary, these results indicate that UFH was highly effective in inhibiting LPS-induced ALI and these protective effects appear to be mediated through inhibition of the proinflammatory NF-κB pathway. Of clinical relevance, UFH may represent a novel strategy for ALI treatment that results from a combination of mechanisms which modulate the entangled processes of coagulation and inflammation.
References
MacLaren, R., and K.A. Stringer. 2007. Emerging role of anticoagulants and fibrinolytics in the treatment of acute respiratory distress syndrome. Pharmacotherapy 27(6): 860–873.
Cepkova, M., and M.A. Matthay. 2006. Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. Journal of Intensive Care Medicine 21: 119–143.
Frutos-Vivar, F., N.D. Ferguson, and A. Esteban. 2006. Epidemiology of acute lung injury and acute respiratory distress syndrome. Seminars in Respiratory and Critical Care Medicine 27: 327–336.
Rubenfeld, G.D., E. Caldwell, E. Peabody, et al. 2005. Incidence and outcomes of acute lung injury. The New England Journal of Medicine 353: 1685–1693.
Ware, L.B., E. Camerer, K.E. Welty-Wolf, et al. 2006. Bench to bedside: targeting coagulation and fibrinolysis in acute lung injury. American Journal of Physiology. Lung Cellular and Molecular Physiology 291(3): L307–L311.
Bastarache, J.A., L.B. Ware, and G.R. Bernard. 2006. The role of the coagulation cascade in the continuum of sepsis and acute lung injury and acute respiratory distress syndrome. Seminars in Respiratory and Critical Care Medicine 27: 365–376.
Schultz, M.J., J.J. Haitsma, H. Zhang, et al. 2006. Pulmonary coagulopathy as a new target in therapeutic studies of acute lung injury or pneumonia: a review. Critical Care Medicine 34: 871–877.
Sapru, A., J.L. Wiemels, J.S. Witte, et al. 2006. Acute lung injury and the coagulation pathway: potential role of gene polymorphisms in the protein C and fibrinolytic pathways. Intensive Care Medicine 32(9): 1293–1303.
Welty-Wolf, K.E., M.S. Carraway, T.L. Ortel, et al. 2006. Blockade of tissue factor–factor X binding attenuates sepsis-induced respiratory and renal failure. American Journal of Physiology. Lung Cellular and Molecular Physiology 290: L21–L31.
Maybauer, M.O., D.M. Maybauer, J.F. Fraser, et al. 2006. Recombinant human activated protein C improves pulmonary function in ovine acute lung injury resulting from smoke inhalation and sepsis. Critical Care Medicine 34(9): 2432–2438.
Yasui, H., E.C. Gabazza, S. Tamaki, et al. 2001. Intratracheal administration of activated protein C inhibits bleomycin-induced lung fibrosis in the mouse. American Journal of Respiratory and Critical Care Medicine 163: 1660–1668.
Uchiba, M., K. Okajima, K. Murakami, et al. 1996. Recombinant thrombomodulin prevents endotoxin-induced lung injury in rats by inhibiting leukocyte activation. American Journal of Physiology 271: L470–L475.
Harada, N., K. Okajima, and M. Uchiba. 2006. Dalteparin, a low molecular weight heparin, attenuates inflammatory responses and reduces ischemia-reperfusion-induced liver injury in rats. Critical Care Medicine 34: 1883–1891.
Yagmurdur, M.C., E. Turk, G. Moray, et al. 2005. Effect of heparin on bacterial translocation and gut epithelial apoptosis after burn injury in the rat: dose-dependent inhibition of the complement cascade. Burns 31: 603–609.
Matzner, Y., G. Marx, R. Drexler, et al. 1984. The inhibitory effect of heparin and related glycosaminoglycans on neutrophil chemotaxis. Thrombosis and Haemostasis 52: 134–137.
Pahl, H.L. 1999. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: 6853–6866.
Zingarelli, B. 2005. Nuclear factor-kappaB. Critical Care Medicine 33: S414–S416.
Liu, S.F., and A.B. Malik. 2006. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. American Journal of Physiology. Lung Cellular and Molecular Physiology 290: L622–L645.
Grisham, M.B., L.A. Hernandez, and D.N. Granger. 1986. Xanthine oxidase and neutrophil infiltration in intestinal ischemia. American Journal of Physiology 251: G567–G574.
Smith, K.M., J.D. Mrozek, S.C. Simonton, et al. 1997. Prolonged partial liquid ventilation using conventional and high-frequency ventilatory techniques: gas exchange and lung pathology in an animal model of respiratory distress syndrome. Critical Care Medicine 25: 1888–1897.
Gando, S. 2010. Microvascular thrombosis and multiple organ dysfunction syndrome. Critical Care Medicine 38(2): S35–S42.
Schwartz, M.D., E.E. Moore, F.A. Moore, et al. 1996. Nuclear factor-kappaB is activated in alveolar macrophages from patients with acute respiratory distress syndrome. Critical Care Medicine 24(8): 1285–1292.
Bhatia, M., and S. Moochhala. 2004. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. The Journal of Pathology 202: 145–156.
Tyrell, D.J., S. Kilfeather, and C.P. Page. 1995. Therapeutic uses of heparin beyond its traditional role as an anticoagulant. Trends in Pharmacological Sciences 16: 198–204.
Morrison, A.M., P. Wang, and I.H. Chaudry. 1996. A novel nonanticoagulant heparin prevents vascular endothelial cell dysfunction during hyperdynamic sepsis. Shock 6: 46–51.
Li, X., Z. Zheng, X. Li, et al. 2012. Unfractionated heparin inhibits lipopolysaccharide-induced inflammatory response through blocking p38 MAPK and NF-κB activation on endothelial cell. Cytokine 60(1): 114–121.
Cinel, I., and R.P. Dellinger. 2007. Advances in pathogenesis and management of sepsis. Current Opinion in Infectious Diseases 20: 345–352.
Li, X., Z. Zheng, Y.R. Mao, et al. 2012. Unfractionated heparin promotes LPS-induced endothelial barrier dysfunction: a preliminary study on the roles of angiopoietin/Tie2 axis. Thrombosis Research 129(5): e223–e228.
Acknowledgments
The authors give special thanks to Dr. Liang Wang (Department of Pathology, China Medical University) for the pathological analysis. The research which discussed unfractionated heparin was supported by the National Natural Science Foundation of China (81101411).
Conflict of Interest
The author declares that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, X., Li, Z., Zheng, Z. et al. Unfractionated Heparin Ameliorates Lipopolysaccharide-Induced Lung Inflammation by Downregulating Nuclear Factor-κB Signaling Pathway. Inflammation 36, 1201–1208 (2013). https://doi.org/10.1007/s10753-013-9656-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-013-9656-5