
Abstract
In Thailand, there are at least ten species of freshwater snails of the family Bithyniidae, most of which can act as intermediate hosts of veterinary and medically important parasites. The genetic variation, geographical distribution, and taxonomic status of some species are obscure, including for Hydrobioides nassa. Thus, this study aims to explore the genetic variation and distribution of H. nassa in north, west, and central Thailand. We collected 264 specimens of H. nassa from 46 localities in 16 provinces using mitochondrial cytochrome c oxidase subunit I (COI) and 16S ribosomal DNA sequences to determine variation. Genetic diversity of H. nassa is relatively high, with 65 and 11 haplotypes of COI and 16S rDNA observed, respectively. The phylogenetic tree and haplotype network analyses classified H. nassa into three haplogroups (haplogroup I–III). Haplogroup I and II belonged to clade A, whereas haplogroup III belonged to clade B. Interestingly, haplogroup III or clade B contained the specimens from Yom river basin in Phrae province, which was the most genetically distinct. Thus, this study suggests that H. nassa in Thailand is a complex of phenotypically similar but genetically distinct species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Freshwater snails, especially the family Bithyniidae are obligate hosts for the immature stages of many trematode parasites (Andrews et al., 2008; Chantima et al., 2013), a number of which are significant pathogens of humans and domestic animals, for example, parasites in the families Opisthorchiidae, Schistosomatidae, and Echinostomatidae (Chantima et al., 2013; Kiatsopit et al., 2014; Kulsantiwong et al., 2015; Namsanor et al., 2015; Kiatsopit et al., 2016; Kulsantiwong et al., 2017). The epidemiological dynamics of trematodes is closely linked to the population ecology and genetics of their snail intermediate hosts (Chitsulo et al., 2000; Chai et al., 2005), which can vary depending on geographical isolation or the environmental conditions present, as evidenced in the liver fluke Opisthorchis viverrini (Poirier, 1886) (Saijuntha et al., 2007). In addition, recent work has shown that even well-recognized snail species can include a number of cryptic species (Kiatsopit et al., 2011), which may have differing host–parasite relationships with specific trematodes (Nadler, 1995; Nadler & DE León, 2011).
The morphology of some snails in the family Bithyniidae is very similar, such as Hydrobioides nassa (Theobald, 1865), Bithynia (syn. Digoniostoma) siamensis siamensis (Morelet, 1866), and Bithynia siamensis goniomphalos (Morelet, 1866). Moreover, these bithyniids coexist with distributions covering large parts of north and central Thailand (Bunchom et al., unpublished). Species identification and differentiation of these snails by morphology are sometimes difficult (Chitramvong, 1992). However, there are several genetic markers that can potentially be used to differentiated these species, such as allozyme markers (Saijuntha et al., 2007; Kiatsopit et al., 2011) and mitochondrial DNA sequences (Kulsantiwong et al., 2013). Thus, using molecular markers to identify and differentiate these snails would be more accurate and reliable, and could also be used for the investigation of genetic variation and molecular systematics (Saijuntha et al., 2007; Kiatsopit et al., 2011; Kulsantiwong et al., 2013).
Hydrobioides nassa has so far been reported from Myanmar, Lao PDR, and Thailand (Brandt, 1974). In Thailand, H. nassa has been found in several areas in the north, west, and central regions of Thailand (Brandt, 1974; Sri-aroon et al., 2007; Kulsantiwong et al., 2013). There are still many areas in Thailand, however, from which it has never been reported or even surveyed. One aim of this study is therefore to determine the geographic distribution of H. nassa in north, west, and central Thailand. The identification of H. nassa, as well as its differentiation from closely related species, for example, Bithynia siamensis, can be determined using mitochondrial cytochrome c oxidase subunit I (COI) sequences. This has been used previously for the differentiation of snails in the Bithyniidae (Kulsantiwong et al., 2013). However, almost all previous reports of genetic variation and population genetic investigations of the Bithyniidae deal only with the genus Bithynia, whereas H. nassa is only poorly understood. Thus, the aim of this study is to determine the genetic variation and population genetics of H. nassa collected from north, west, and central Thailand using mitochondrial COI and 16S ribosomal DNA sequences.
Materials and methods
Specimen collection
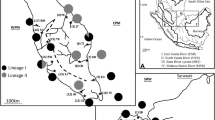
The 264 specimens of H. nassa (Fig. 1) were collected from 46 localities that belonged to seven river basins in 16 provinces in north, west, and central Thailand (Table 1; Fig. 2). Snails were collected from natural habitats, for example, paddy fields, small streams, clay pools, and flowing water near roadsides, by handpicking and scooping. Snails were cleaned with tap water several times, dried with tissue paper, kept in newspaper, and transported to the laboratory. The snails were then identified by standard morphological criteria described by Brandt (1974), Upatham et al. (1983), and Chitramvong (1992). The shell is small (length ~ 5.05 mm, width ~ 2.84 mm), with both transverse growth lines and spiral lines on the shell surface. The apex of the shell is usually not eroded, but occasionally old shells are found with apices a little eroded. The umbilicus is completely closed. The outer part of the last whorl is extended as a flange that causes the outer (palatal) lip to be sinuous in shape. The aperture of the shell also has a weak incision (indentation) at the middle of the basal lip (Fig. 1).

Morphology of the shell and operculum of Hydrobioides nassa

Map of localities for sample collection related to seven river basins in north, west, and central Thailand
After identification, the head and foot were removed and kept in 80% alcohol for further DNA analysis. The shell and operculum were air dried and deposited in the Walai Rukavej Botanical Research Institute, Mahasarakham. The ethics of animal use and biosafety for this research were approved by the Animal Care and Use Committee (permission number 008/2561) and the Institute Biosafety Committee (Permission Number 044/2561) of Thammasat University, respectively.
Molecular analyses
DNA was individually extracted from the head and foot of snails using the E.Z.N.A.® Mollusc DNA kit (Omega bio-tek, USA) following manufacturing protocol. To amplify the COI region, the polymerase chain reaction (PCR) was performed using primers LCO1490 (5′-GGT CAA CAA ATC ATA AAG ATA TTG G-3′) and HCO2198 (5′-TAA ACT TCA GGG TGA CCA AAA AAT CA-3′) (Folmer et al., 1994), whereas 16S rDNA was amplified by using primers 16sar-L (5′-CGC CTG TTT ATC AAA AAC AT-3′) and 16sbr-H (5′-CCG GTC TGA ACT CAG ATC ACG T-3′) (Palumbi et al., 2002). The PCR mix of 25 µl contained 18.8 µl of distilled water, 2.5 µl of 10 × Ex PCR buffer, 0.5 µl of 10 µM dNTPs, 1 µl of each primer (10 µM), 0.2 µl of 5 U/ul Ex Taq polymerase (Takara, Japan), and 1 µl of DNA template (~ 10 to 50 ng). The PCR condition to amplify both regions was the same as follows: initial denaturation at 94°C for 5 min, followed by 35 cycles of 95°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 30 s, after which, a final extension was carried out at 72°C for 8 min. The PCR products were separated on 1% agarose gel. The amplicon sizes with approximately 650 and 470 bp of the COI and 16S rDNA regions, respectively, were cut and purified by using E.Z.N.A.® Gel Extraction kit (Omega bio-tek, USA). DNA sequencing was performed at Eurofins genomics company, Japan.
Data analyses
The DNA sequences were assembled and edited using BioEdit v.7.2.6 (Hall, 1999). The 195 COI sequences of the Bithyniidae were determined by Kulsantiwong et al. (2013) and deposited in GenBank, namely B. s. goniomphalos (KY118603–KY18632), B. s. siamensis (KY118633–KY118672), Bithynia funiculata (Walker, 1972) (KY118590–KY118596 and KY118598–KY118602), H. nassa (KY118743–KY118748 and KY118751–KY118765), Wattebledia baschi (Brandt, 1968) (KY118766–KY118771), Wattebledia crosseana (Wattebled, 1886) (KY118776, KY118785–KY118786, KY118788–KY118790, and KY118794–KY118798) Wattebledia siamensis (Moellendorff, 1902) (KY118799–KY11806), Gabbia wykoffi (Brandt, 1968) (KY118684–KY118731 and KY118733–KY118742), Gabbia pygmaea (Preston, 1908) (KY118681–KY118683), Gabbia erawanensis (Prayoonhong, Chitramvong & Upatham, 1990) (KY118673–KY118680). These were included in our analysis. The COI and 16S rDNA sequences were multiply aligned using Clustal W (Larkin et al., 2007).
All COI sequences obtained in this study were blasted in GenBank (https://blast.ncbi.nlm.nih.gov/Blast.cgi), Barcode of Life Data Systems (http://www.boldsystems.org), and the bPTP server (https://species.h-its.org/) for species confirmation (Zhang et al., 2013). The phylogenetic analyses were constructed based on the neighbor-joining method (Saitou & Nei, 1987) using the Kimura 2-parameter model (Kimura, 1980) with bootstrap support of 1,000 replications, whereas maximum likelihood was run by using the general time reversible with gamma distribution model (GTR + G + I model) (Nei & Kumar, 2000), with bootstrap support of 1,000 replications using MEGA X program (Kumar et al., 2018). The Hasegawa–Yano–Kishino model incorporating invariant sites and a gamma distribution (HYK + G + I) model was selected for Bayesian interference (BI) analyses as determined by MrModelTest v.2.3 (Nylander, 2008). The BI analysis was run using MrBayes v.3.2 (Ronquist et al., 2012) with 10,000,000 generations using four chains. The trees were sampled every 100th generation resulting in 100,000 trees. The first 73,500 trees were excluded as a burn-in because the standard deviation of split frequencies (ASDF) was above 0.01. Furthermore, the convergence of the run was assessed by checking the potential scale reduction factor (PSRF) values of each parameter in MrBayes and Effective Sample Size (ESS) values of each parameter in Tracer 1.6 (Rambaut et al., 2014). A value of PSRF close to 1.00 and that of ESS above 200 were considered as good indicators of convergence. Considering the results of checking these values, the remaining trees (26,500 trees) were used to calculate Bayesian posterior probabilities of individual clades. Then, the BI tree was visualized in FigTree v1.4.2 (Rambaut, 2012). A median joining haplotype network (Bandelt et al., 1999) was generated using Network v.5.0 (http://www.fluxus-engineering.com). The genetic diversity indices, i.e., haplotype diversity (Hd) and nucleotide diversity (π), were performed in DnaSp v.5.10.01 (Librado & Rozas, 2009). Pairwise genetic differences (ФST) based on the Kimura-2-parameter model (Kimura, 1980) were calculated in Arlequin v.3.5.2.2 (Excoffier & Lischer, 2010), and Nei’s genetic distance (Nei, 1978) was calculated in the R program (R Core Team, 2013).
Results
A total of 264 and 205 specimens of H. nassa were analyzed for COI and 16S rDNA sequences, respectively. All nucleotide sequences generated in this study were submitted to GenBank under the accession numbers MK639930–MK640193 and MK629019–MK629223 for COI and 16S rDNA, respectively. The COI sequences generated in this study were mostly similar with H. nassa in the GenBank database, Barcode of Life Data Systems, and bPTP server at 98–100%. Comparison 658 bp of COI sequences revealed that the 98 variable positions found consisted of 59 transitions, 30 transversions, and 9 multiple mutations (Table 2), which were classified into 65 haplotypes (H1–H65). These nucleotide variations translated into different amino acid sequences (Table 2). The haplotype diversity (Hd) and nucleotide diversity (π) of COI were 0.862 ± 0.018 and 0.0147 ± 0.0013, respectively (Table 3). The comparison of the 492 bp sequence of 16S rDNA sequences revealed 12 variable positions, consisting of 8 transitions, 3 transversions and 1 multiple mutation (data not shown), which were classified into 11 haplotypes (H1–H11). The haplotype diversity and nucleotide diversity of 16S rDNA were 0.502 ± 0.039 and 0.0025 ± 0.0003, respectively (Table 3). Haplotype H1 is the most common for both COI and 16S rDNA shared between different populations from different river basins. However, all river basins have unique/specific haplotypes (Table 3; Fig. 3). Pairwise genetic differences (ФST) between the populations from seven river basins, namely Kok, Ping, Wang, Yom, Nan, Mekong, and Chao Phraya ranged between 0.038 and 0.239 and 0.000 and 0.675 for COI and 16S rDNA analyses, respectively (Table 4). Significant genetic differences for COI were observed between all populations from the different river basins, except the populations from the Kok vs Wang, and Kok vs Mekong river basins (Table 4). Whereas 16S rDNA showed several nonsignificant genetic differences, i.e., the populations from Ping vs Wang, Nan, and Chao Phraya; Wang vs Nan and Chao Phraya; and Nan vs Chao Phraya river basins (Table 4).

Maximum likelihood tree of the Bithyniidae in Thailand constructed based on COI sequences combining the minimum spanning haplotype networks of H. nassa generated from COI and 16S rDNA sequences. A monophyletic group of H. nassa was classified into two clades (A and B): clade A contained two haplogroups (I and II), and clade B contained haplogroup III. Nodal supports are sequential values of bootstrap values generated by neighbor-joining and maximum likelihood analyses, and Bayesian posterior probability generated by BI analysis
A minimum spanning haplotype network constructed from COI and 16S rDNA sequences also demonstrated that three haplogroups (I–III) could be classified (Fig. 3). Haplogroup I was the largest group, containing haplotypes from all seven river basins. It differed from haplogroup II at five and one mutational steps for COI and 16S rDNA, respectively. Haplogroup II contained haplotypes from the Kok, Ping, Yom, Nan, and Mekong river basins. Haplogroup III contained eight haplotypes and belonged to the Yom river basin in Phrae province. This was the most distinct group, differing from the other at 25 and five mutational steps for COI and 16S rDNA, respectively (Fig. 3). The phylogenetic tree constructed using COI sequences of the Bithyniidae revealed that four genera, namely Bithynia, Gabbia, Wattebledia, and Hydrobioides, were clearly separated (Fig. 3). All specimens from our study were grouped in the monophyletic group of the genus Hydrobioides (Fig. 3). A monophyletic group of H. nassa was classified into two difference clades (A and B), with clade A contained two different haplogroups, namely haplogroup I and haplogroup II, whereas clade B contained haplogroup III.
Genetic distance analysis of the bithyniids (except genus Gabbia) revealed that Nei’s genetic distance comparison within species/subspecies ranged between 0.014–0.142, between species 0.214–0.386, and between genera 0.461–0.705 (Table 5). Comparison between haplogroups I and II showed a level of genetic distance at 0.014, whereas haplogroup III was distinct from haplogroups I and II at 0.142 and 0.135, respectively. Interestingly, the genetic distance of H. nassa haplogroup III from the others was greater than the distance between the two subspecies of Bithynia siamensis, namely B. s. goniomphalos and B. s. siamensis at 0.072, but lower than the genetic distance between species, 0.214–0.386 (Table 5). Moreover, pairwise genetic differentiation (ФST) showed significant differences between all haplogroups of H. nassa, as well as between subspecies, species, and genera of the bithyniids (Table 5).
Discussion
This study provides the first report on the genetic diversity and population genetics of H. nassa in Thailand. Based on current molecular analyses, broader geographical ranges of H. nassa were observed in Lamphun, Phrae, Phayao, Uttaradit, and Nan provinces in the north; Tak province in the west; Nakhon Sawan, Sukhothai, Phitsanulok, Kamphaeng Phet, and Lop Buri provinces in the central regions of Thailand compared to previous reports (Brandt, 1974; Sri-aroon et al., 2007; Kulsantiwong et al., 2013). In addition, H. nassa coexists with a phenotypically similar species, i.e., Bithynia siamensis in several areas in this study (Bunchom et al., unpublished). This may lead to confusion and misidentification if morphology alone is used as the identifying criterion. This study shows that using the COI sequence for the identification of H. nassa, as well as differentiation from a phenotypically similar species, B. siamensis, is both precise and reliable.
Previously, the only genetic variation found in freshwater snails belonging the family Bithyniidae has been reported for Bithynia siamensis using allozyme electrophoresis (Saijuntha et al., 2007; Kiatsopit et al., 2011, 2013) and mitochondrial COI sequences (Kulsantiwong et al., 2013), whereas microsatellite markers have been used for the population genetic analysis of Bithynia tentaculata (Linnaeus, 1758) in North America and Europe (Perez et al., 2016). These demonstrated high levels of genetic variation in the bithyniids, as revealed in H. nassa in this study. Two clades and three haplogroups were classified based on both COI and 16S rDNA analyses. Based on COI sequences analysis, genetic differences (ФST) were significantly observed (P value < 0.05) between the populations of H. nassa from several different river basins. Thus, gene flow within H. nassa in Thailand would be possibly influenced by flooding or the flow pattern of the main rivers in a particular region. The other supporting evidence that B. s. goniomphalos shows genetic differences and clustering correlated to river wetland systems in the northeast Thailand and Lao PDR was found by multilocus enzyme electrophoresis (Saijuntha et al., 2007; Kiatsopit el al., 2013). However, nonsignificant genetic differences between some populations of adjacent/connected river basins were observed. This shows that gene flow/migration between these populations has been occurring—for example, the populations from the Kok and Wang, Kok, and Mekong river basins.
Moreover, northern Thailand is geographically characterized by several mountain ranges, which can act as natural barriers blocking the migration/gene flow of these snails, as evidenced in other terrestrial, pulmonate snails in this region (Prasankok & Panha, 2011). Our study demonstrated that clade B (haplogroup III) of H. nassa—located in the Phrae–Nan–Uttaradit cordillera and surrounded by high mountain ranges—was the most genetically distinct. It is likely that the limited gene flow and migration of H. nassa in this area led to local adaptation with high genetic differences. This phenomenon has been found previously in the other organisms in this area, such as the black fly, Simulium weji Takaoka, 2001 (Pramual & Pangjanda, 2015), catfish, Clarias macrocephalus Günther, 1864 (Na-Nakorn et al., 2004), and the Tokay gecko, Gekko gecko (Linnaeus, 1758) (Saijuntha et al., 2019).
Our current findings suggest that H. nassa is a relatively genetically diverse species and may consist of a complex of phenotypically similar but genetically distinct species. However, this requires a further intensive study of the phenotypic and genetic variability of H. nassa extended to more geographical areas in Thailand, as well as other countries in Southeast Asia, to determine the taxonomic status and genetic structure of this snail throughout its distributional range.
References
Andrews, R. H., P. Sithithaworn & T. N. Petney, 2008. Opisthorchis viverrini: an underestimated parasite in world health. Trends in Parasitology 24: 497–501.
Bandelt, H. J., P. Forster & A. Rohl, 1999. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16: 37–48.
Brandt, R. A. M., 1974. The non-marine aquatic Mollusca of Thailand. Arch Mollusken 105: 1–423.
Chai, J. Y., K. D. Murrell & A. J. Lymbery, 2005. Fish-borne parasitic zoonoses: status and issues. International Journal for Parasitology 35: 1233–1254.
Chantima, K., J. Y. Chai & C. Wongsawad, 2013. Echinostoma revolutum: freshwater snails as the second intermediate hosts in Chiang Mai, Thailand. The Korean Journal of Parasitology 51: 183–189.
Chitramvong, Y. P., 1992. The Bithyniidae (Gastropoda: Prosobranchia) of Thailand: comparative external morphology. Malacological Review 25: 38.
Chitsulo, L., D. Engels, A. Montresor & L. Savioli, 2000. The global status of schistosomiasis and its control. Acta Tropica 77: 41–51.
Excoffier, L. & H. E. Lischer, 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10: 564–567.
Folmer, O., M. Black, W. Hoeh, R. Lutz & R. Vrijenhoek, 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology 3: 294–299.
Hall, T. A., 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series. Information Retrieval Ltd., London: 1979–2000.
Kiatsopit, N., P. Sithithaworn, T. Boonmars, S. Tesana, A. Chanawong, W. Saijuntha, T. N. Petney & R. H. Andrews, 2011. Genetic markers for studies on the systematics and population genetics of snails, Bithynia spp., the first intermediate hosts of Opisthorchis viverrini in Thailand. Acta Tropica 118: 136–141.
Kiatsopit, N., P. Sithithaworn, W. Saijuntha, T. N. Petney & R. H. Andrews, 2013. Opisthorchis viverrini: Implications of the systematics of first intermediate hosts, Bithynia snail species in Thailand and Lao PDR. Infection, Genetics and Evolution 14: 313–319.
Kiatsopit, N., P. Sithithaworn, K. Kopolrat, R. H. Andrews & T. N. Petney, 2014. Seasonal cercarial emergence patterns of Opisthorchis viverrini infecting Bithynia siamensis goniomphalos from Vientiane Province, Lao PDR. Parasites & Vectors 7: 551.
Kiatsopit, N., P. Sithithaworn, K. Kopolrat, J. Namsanor, R. H. Andrews & T. N. Petney, 2016. Trematode diversity in the freshwater snail Bithynia siamensis goniomphalos sensu lato from Thailand and Lao PDR. Journal of Helminthology 90: 312–320.
Kimura, M., 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 16: 111–120.
Kulsantiwong, J., S. Prasopdee, J. Ruangsittichai, W. Ruangjirachuporn, T. Boonmars, V. Viyanant, P. Pierossi, P. D. Hebert & S. Tesana, 2013. DNA barcode identification of freshwater snails in the family Bithyniidae from Thailand. PLoS ONE 8: e79144.
Kulsantiwong, J., S. Prasopdee, S. Piratae, P. Khampoosa, C. Thammasiri, A. Suwannatrai, T. Boonmars, V. Viyanant, J. Ruangsitichai, P. Tarbsripair & S. Tesana, 2015. Trematode infection of freshwater snail, family Bithyniidae in Thailand. Southeast Asian Journal of Tropical Medicine Public Health 46: 396–405.
Kulsantiwong, J., S. Prasopdee, N. Labbunruang, M. Chaiyasaeng & S. Tesana, 2017. Habitats and trematode infection of Bithynia siamensis gonimphalos in Udon Thani province, Thailand. Southeast Asian Journal of Tropical Medicine and Public Health 48: 975–982.
Kumar, S., G. Stecher, M. Li, C. Knyaz & K. Tamura, 2018. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution 35: 1547–1549.
Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson & D. G. Higgins, 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948.
Librado, P. & J. Rozas, 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Nadler, S. A., 1995. Microevolution and the genetic structure of parasite populations. The Journal of Parasitology 81: 395–403.
Nadler, S. A. & G. P. DE Leon, 2011. Integrating molecular and morphological approaches for characterizing parasite cryptic species: implications for parasitology. Parasitology 138: 1688–1709.
Namsanor, J., P. Sithithaworn, K. Kopolrat, N. Kiatsopit, O. Pitaksakulrat, S. Tesana, R. H. Andrews & T. N. Petney, 2015. Seasonal transmission of Opisthorchis viverrini sensu lato and a Lecithodendriid trematode species in Bithynia siamensis goniomphalos snails in Northeast Thailand. The American Journal of Tropical Medicine and Hygiene 93: 87–93.
Na-Nakorn, U., W. Kamonrat & T. Ngamsiri, 2004. Genetic diversity of walking catfish, Clarias macrocephalus, in Thailand and evidence of genetic introgression from introduced farmed C. gariepinus. Aquaculture 240: 145–163.
Nei, M., 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 23: 341–369.
Nei, M. & S. Kumar, 2000. Molecular Evolution and Phylogenetics. Oxford University Press, New York.
Nylander, J. A. A., 2008. MrModeltest v2.3 Program Distributed by the Author. Evolutionary Biology Centre, Uppsala University.
Palumbi, S. R., A. P. Martin, S. Romano, W. O. MacMillan, L. Stice & G. Grabowski, 2002. The Simple Fool’s Guide to PCR version 2.0. Department of Zoology and Kewalo Marine Laboratory, University of Hawaii, Honolulu.
Perez, K. E., R. L. Werren, C. A. Lynum, L. A. Hartman, G. Majores & R. A. Cole, 2016. Genetic structure of faucet snail, Bithynia tentaculata populations in North America, based on microsatellite markers. Freshwater Mollusk Biology and Conservation 19: 56–69.
Pramual, P. & S. Pangjanda, 2015. Effects of habitat specialization on population genetic structure of black fly Simulium weji Takaoka (Diptera: Simuliidae). Journal of Asia-Pacific Entomology 18: 33–37.
Prasankok, P. & S. Panha, 2011. Genetic structure of the common terrestrial pulmonate snail, Cryptozona siamensis (Pfeiffer, 1856), in Thailand. Biochemical Systematics and Ecology 39: 449–457.
R Core Team, 2013. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
Rambaut, A., 2012. FigTree v.1.4.2: Tree Figure Drawing Tool. Available online at: http://tree.bio.ed.ac.uk/software/figtree
Rambaut, A., M. A. Suchard, D. Xie & A. J. Drummond, 2014. Tracer v1.6. Available online at: http://beast.bio.ed.ac.uk/Tracer
Ronquist, F., M. Teslenko, P. van der Mark, D. L. Ayres, A. Darling, S. Höhna, B. Larget, L. Liu, M. A. Suchard & J. P. Huelsenbeck, 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542.
Saijuntha, W., P. Sithithaworn, S. Wongkham, T. Laha, V. Pipitgool, S. Tesana, N. B. Chilton, T. N. Petney & R. H. Andrews, 2007. Evidence of a species complex within the food-borne trematode Opisthorchis viverrini and possible co-evolution with their first intermediate hosts. International Journal for Parasitology 37: 695–703.
Saijuntha, W., S. Sedlak, T. Agatsuma, K. Jongsomchai, W. Pilap, W. Kongbuntad, W. Tawong, W. Suksavate, T. N. Petney & C. Tantrawatpan, 2019. Genetic structure of the red-spotted tokay gecko, Gekko gecko Linnaeus, 1758 (Squamata: Gekkonidae) in mainland Southeast Asia. Asian Herpetological Research. https://doi.org/10.16373/j.cnki.ahr.180066.
Saitou, N. & M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4: 406–425.
Sri-aroon, P., P. Butraporn, J. Limsoomboon, M. Kaewpoolsri, Y. Chusongsang, P. Charoenjai, P. Chusongsang, S. Numnuan & S. Kiatsiri, 2007. Freshwater mollusks at designated areas in eleven provinces of Thailand according to the water resource development projects. Southeast Asian Journal of Tropical Medicine and Public Health 38: 294–301.
Upatham, E. S., S. Sornmani, V. Kitikoon, C. Lohachit & J. B. Burch, 1983. Identification key for the fresh- and brackish-water snails of Thailand. Malacological Review 16: 107–132.
Zhang, J., P. Kapli, P. Pavlidis & A. Stamatakis, 2013. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29: 2869–2876.
Acknowledgements
This study was supported by the Thailand Research Fund through the Royal Golden Jubilee Ph.D. program (Grant No. PHD/0095/2558) to Naruemon Bunchom. Thammasat University financial supports this project to Chairat Tantrawatpan. Takeshi Agatsuma was supported by Grants-in-Aid for Scientific Research (B) (26305011). The authors would like to thank the Mahasarakham University for financially supporting Weerachai Saijuntha, enabling him to present this work in the first FMCS International Freshwater Mollusk Meeting 2018, Verbania, Italy. The authors would also like to thank Dr. Wittaya Tawong for BI analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Guest editors: Manuel P. M. Lopes-Lima, Nicoletta Riccardi, Maria Urbanska & Ronaldo G. Sousa / Biology and Conservation of Freshwater Molluscs
Rights and permissions
About this article

Cite this article
Bunchom, N., Saijuntha, W., Pilap, W. et al. Genetic variation of a freshwater snail Hydrobioides nassa (Gastropoda: Bithyniidae) in Thailand examined by mitochondrial DNA sequences. Hydrobiologia 848, 2965–2976 (2021). https://doi.org/10.1007/s10750-019-04013-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-019-04013-2