
Abstract
Indoplanorbis exustus, a freshwater pulmonate snail, is widely distributed in tropical and subtropical zones and plays a significant role as an intermediate host for trematode parasites. Various genetic markers have been used for species identification and phylogenetic studies of this snail. However, there are limited studies about their molecular genetics based on nuclear ribosomal DNA (rDNA) genes. A genetic analysis of I. exustus in Thailand was conducted based on the nuclear 18S rDNA (339 bp) and 28S rDNA (1036 bp) genes. Indoplanorbis snails were collected from 29 localities in 21 provinces covering six regions of Thailand. Nucleotide sequences from 44 snails together with sequences from the GenBank database were examined for phylogenetic relationships and genetic diversity. All sequences of the selected nucleotide regions exhibited a high level of similarity (99%) to the sequences of I. exustus in the GenBank database. The maximum likelihood tree based on the 18S and 28S rDNA fragment sequences of I. exustus in Thailand revealed only one group with clear separation from another genus in the family Planorbidae. The I. exustus 28S rDNA sequences showed intraspecific genetic divergence ranging from 0 to 0.78% and were classified into 8 different haplotypes. Conversely, the 18S rDNA data showed lower variation than the 28S rDNA data and revealed a single haplotype and intraspecific distances of zero among all sampled individuals. The haplotype network of 28S rDNA sequences of I. exustus in Thailand revealed six unique haplotypes and two haplotypes shared by at least two regions. Overall, both markers were successful in the identification of I. exustus. However, these markers, particularly the 18S rDNA, may not be suitable for genetic analysis within the species, particularly for population genetic studies, due to their limited variation as seen in this study. In summary, this study not only enhances understanding of genetic variation in I. exustus but is also useful for the selection of molecular markers in future genetic research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Indoplanorbis exustus (Deshayes 1834) is a freshwater pulmonate snail belonging to the family Planorbidae that is most widespread in tropical and subtropical zones (Morgan et al. 2002; Mouahid et al. 2018). This snail is widely distributed across tropical Asia, throughout mainland Southeast Asia, southern China, the Indian subcontinent, Central Asia, and West Asia (Gauffre-Autelin et al. 2017; Liu et al. 2010; Mouahid et al. 2018; Sil et al. 2022). There are also reports of I. exustus that have been introduced from different parts of the world, such as French Polynesia, French West Indies, Ivory Coast, Nigeria, Japan, and Yemen (Ikeda et al. 2021; Kristensen and Ogunnowo 1987; Meyer et al. 2021; Mouahid et al. 2018; Mouchet et al. 1987; Pagad 2020). In addition, this species has been listed as an alien species to be given top national quarantine significance in the USA, where it is considered a serious threat that could negatively affect agriculture, ecosystems, human health, and commerce (Cowie et al. 2009).
I. exustus plays an important role as an intermediate host for several trematode parasites (Ardpairin et al. 2022; Krailas et al. 2022; Wiroonpan et al. 2021). The widespread invasion and environmental tolerance of I. exustus make it important in veterinary and medical sciences (Devkota et al. 2015). In Thailand, I. exustus is widely distributed in various freshwater habitats, such as canals, swamps, rivers, and paddy fields (Saijuntha et al. 2021). It serves as an intermediate host for Schistosoma spindale and S. indicum, which cause schistosomiasis in buffalo and dairy cattle and can also cause cercarial dermatitis in humans (Krailas et al. 2022; Nithiuthai et al. 2004). In addition, this snail serves as the first intermediate host of Echinostoma spiniferum, Clinostomum giganticum, Echinostoma malayanum and Petasiger exaeretus, which are trematode parasites of humans and nonhuman animals (Chai 2019; Krailas et al. 2022; Wiroonpan et al. 2021; Won et al. 2020).
The transmission and dispersal of trematodes are closely related to snail intermediate hosts; thus, accurate identification of snail species is paramount for understanding the epidemiology of parasites and focal control in endemic areas (Maes et al. 2022). Planorbidae snails have a highly variable shell morphology, which is associated with ecological phenotypic plasticity (Andrus et al. 2023). The shell morphology of I. exustus is very similar to that of other Planorbidae snails, such as Planorbella duryi (Helisoma duryi) (Wetherby 1879) and Biomphalaria pfeifferi (Krauss 1848); thus, an examination of the characteristics of the reproductive system is required to distinguish among them (Kristensen and Ogunnowo 1987). As a result, the identification of I. exustus by only shell morphological characteristics may lead to misidentification.
Certainly, molecular methods, specifically those using DNA sequences, have been widely adopted by malacologists to identify species. Molecular data provide a reliable and helpful source of taxonomic and phylogenetic information, which can be used to correct taxonomic errors due to within- or between-species morphological variation (Dumidae et al. 2021;Jørgensen et al. 2011 ; Mouahid et al. 2018). To date, molecular-based identification using molecular genetic markers has been widely used to aid species delimitation in Planorbidae snails (Jørgensen et al. 2011; Mouahid et al. 2018). Both mitochondrial and nuclear gene sequences have been used for molecular identification and phylogenetic analyses of I. exustus (Mouahid et al. 2018; Saijuntha et al. 2021). The mitochondrial genes include cytochrome c oxidase subunit I (COI) and 16S rDNA (Gauffre-Autelin et al. 2017; Liu et al. 2010; Mouahid et al. 2018; Saijuntha et al. 2021). The nuclear genes include the 18S rDNA, 28S rDNA, and internal transcribed spacer (ITS) regions (Jørgensen et al. 2011; Mouahid et al. 2018). Different molecular markers may yield different genetic results (Jørgensen et al. 2011; Mouahid et al. 2018).
Nucleotide sequences of the mitochondrial and nuclear genes have been deposited in GenBank. Among the I. exustus sequences collected around the world, nucleotide sequences are available in GenBank for the COI gene (374 entries), 16S rDNA (230), 18S rDNA (33), 28S rDNA (4), and ITS (185) (retrieved 5 April 2023). It is evident that there have been few studies of I. exustus based on 18S and 28S rDNA sequences. To the best of our knowledge, there is only a single entry for the 18S rDNA (GenBank accession no. AY282598) and 28S rDNA (GenBank accession no. AF435662) genes of I. exustus from Thailand. However, the 18S and 28S rDNA genes have been utilized for species delineation in the family Planorbidae (Jørgensen et al. 2011; Morgan et al. 2002; Sil et al. 2022). There have been no reports on the genetic variation in Thai I. exustus strains based on these genetic markers.
Many previous genetic variation studies of I. exustus in Thailand focused exclusively on the COI and 16S rDNA genes. Comparable 18S and 28S rDNA studies have not been performed in I. exustus. To obtain more accurate genetic information on I. exustus populations, more molecular markers and more specimens are needed. Therefore, the aim of this study was to identify and analyze the genetic variation in I. exustus collected from Thailand using nuclear 18S and 28S ribosomal DNA sequences.
Materials and methods
Ethical approval
In this study, the experiments involving invertebrate animals (snails) received approval from the Center for Animal Research at Naresuan University (Project Ethics No: NU-AQ640803). All procedures for the sampling and euthanasia of snails were performed following the American Veterinary Medical Association (AVMA) (2020) and ARRIVE guidelines. Additionally, the biosafety protocols were approved by the Naresuan University Institutional Biosafety Committee (Project No: NUIBC MI 64-09-34).
Sample collection and species identification
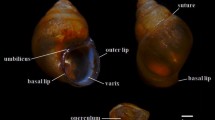
Samples of I. exustus were randomly collected with wire-mesh scoops or by hand in 2020 and 2022 from 29 localities in 21 provinces in North, Northeast, Central, West, East, and South Thailand (Table 1, Fig. S1). No specific permission is required to collect this snail. The snail samples in this study were collected mostly from paddy fields, ponds, and canals and transported to the laboratory. All snails were cleaned with tap water and dried with tissue paper, their shells were photographed, and they were measured for shell width and length. The snails were then primarily identified based on the shell morphological criteria described by Brandt (Brandt 1974) and Frandsen (Frandsen 1983). Briefly, I. exustus has a discoid shell with a dorso-ventrally flattened shape and rapidly increasing whorls. Each whorl is taller than it is wide. The aperture is expanded, and the peristome is sharp without a lip (Fig. 1) (Brandt 1974; Frandsen 1983). After identification, the snails were dissected to remove the soft body, and a piece of foot tissue measuring approximately 25 mg was cut from each individual and preserved at −20 °C for further DNA extraction.

Shell morphology of I. exustus collected in the field in Thailand
Genomic DNA extraction
DNA was extracted from the snail tissue using the NucleoSpin® Tissue Kit (Macherey-Nagel, Duren, Germany) following the manufacturer’s instructions. The quality of the genomic DNA was checked by running it on a 0.8% agarose gel in 1 × TBE buffer at 100 V for 35 min. The gel was stained with ethidium bromide for 8 min, destained with distilled water for 15 min, and photographed under UV light. The genomic DNA was stored at −20 °C prior to further processing.
Polymerase chain reaction (PCR) and sequencing
To amplify partial fragments of the 18S and 28S rDNA of the snails, polymerase chain reaction (PCR) was performed. A list of primers and the PCR cycling profiles are presented in Table 2. The PCR volume was 30 μl, consisting of 15 μl of OnePCR Ultra (Biohelix, New Taipei, Taiwan), 1.5 μl of each primer at 5 μM (0.25 μM), 9 μl of distilled water, and 3 μl of the DNA template (20–200 ng). The PCR products were examined by 1.2% agarose gel electrophoresis in 10X TBE buffer and run at 100 V for 35 min. The agarose gel containing DNA fragments was stained with ethidium bromide and visualized by ultraviolet light. Amplified products were purified using a NucleoSpin® Gel and PCR Clean-Up Kit (Macherey-Nagel, Germany) following the manufacturer’s recommendations. Purified DNA products were checked with a 1.2% agarose gel. DNA sequencing was conducted at Macrogen Inc., Seoul, Korea.
18S rDNA and 28S rDNA sequences from GenBank
In this study, we included additional I. exustus sequences (4 sequences) as well as sequences from 15 genera (41 sequences) within the family Planorbidae, which were downloaded from GenBank. The sequences of Radix auricularia (Lymnaeidae) and Physa acuta (Physidae) were employed as outgroup for reconstructing the phylogeny because their morphologies, physiologies, and ecologies were similar to Planorbidae (Jørgensen et al. 2011).
Sequencing and phylogenetic analysis
Forward and reverse DNA sequences were assembled and edited using SeqMan II (DNASTAR, Madison, WI, USA). These sequences, along with reference sequences from the NCBI database, were aligned by using Clustal W in MEGA 7 software (Kumar et al. 2016). The nuclear 18S and 28S rDNA sequences were blasted against the GenBank database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to confirm the species identity of I. exustus.
Phylogenetic trees were constructed via the maximum likelihood (ML) and neighbor joining (NJ) methods in MEGA 7 software (Kumar et al. 2016). Bayesian information criterion scores were used for selecting the best model for analysis of phylogeny. ML phylogenetic analysis was carried out based on the Tamura 3-parameter model (Tamura 1992). NJ was performed using the Kimura 2-parameter model (Kimura 1980) with 1000 bootstrap (BS) iterations for tree topology support. The two methods revealed congruent topologies; hence, we present the maximum likelihood (ML) tree with bootstrap values derived from both the ML and NJ methods.
Genetic analysis
The observed number of haplotypes, segregating sites, and haplotype frequencies were estimated using the DnaSP program, version 5 (Librado and Rozas 2009). Genealogical relationships between haplotypes were further elucidated with a median-joining (MJ) network algorithm (Bandelt et al. 1999) using PopART v1.7 (Leigh and Bryant 2015). The p-distances between haplotypes were estimated using MEGA version 7.0 software to assess the level of genetic variation.
Results and discussions
Molecular identification of Indoplanorbis exustus
The molecular identification of I. exustus (44 sequences) based on the 18S and 28S rDNA genes was congruent with the morphological identification. Based on the 339 bp of the 18S rDNA gene, all 44 sequences (GenBank accession nos. OQ975759–OQ975802) in this study showed the highest similarity (100%) with the known sequence of I. exustus from Thailand (GenBank accession no. AY282598). Additionally, the 28S rDNA sequence (1036 bp) of I. exustus (GenBank accession nos. OQ975465-OQ975508) had a similarity of 100% with another from I. exustus in Thailand (GenBank accession no. AF435662). This confirmed the identity of the snails in our study as I. exustus.
I. exustus is widely distributed in Thailand and plays an important role as an intermediate host for medically and veterinary important trematodes (Bawm et al. 2022; Devkota et al. 2015; Krailas et al. 2022; Saijuntha et al. 2021). The phylogenetic analysis of I. exustus based on the mitochondrial DNA sequences indicates that this snail may belong to a cryptic species complex or comprise more than one species, which cannot be distinguished by morphological characteristics (Gauffre-Autelin et al. 2017; Liu et al. 2010; Mouahid et al. 2018; Saijuntha et al. 2021). Sometimes, the identification of a species with only one marker can lead to misidentification (Vences et al. 2005). We have identified I. exustus based on its nuclear 18S and 28S rDNA genes. All I. exustus sequences were compared with NCBI BLAST results. Our results showed the highest similarity (100%) with the sequences of I. exustus in GenBank for both the 18S and 28S rDNA genes. This confirms that the 18S and 28S rDNA genes are sufficiently effective for identifying I. exustus, since they are highly conserved within species (Hwang and Kim 1999). Our findings were in concordance with those from previous studies (Jørgensen et al. 2011; Morgan et al. 2002). In addition, the 18S and 28S rDNA genes were successful in the classification of other snails in the family Planorbidae, such as Bulinus spp., Ceratophallus spp., Drepanotrema spp., Gyraulus spp., Helisoma spp., Planorbis planorbis, Planorbarius corneus, and Planorbella duryi (Jørgensen et al. 2011; Morgan et al. 2002). This emphasizes that 18S and 28S rDNA markers are sufficient for delimitation up to the genus or species level within the family Planorbidae.
Phylogenetic analyses
Molecular phylogenetic analyses of I. exustus were conducted based on 18S and 28S rDNA sequences, revealing consistent results. The phylogenetic tree showed that the group of I. exustus was clearly separated from other snail genera in the family Planorbidae. Both methods (ML and NJ) of phylogenetic tree construction based on the 44 sequences of 18S rDNA (339 bp) in the present study along with 3 sequences downloaded from GenBank revealed only one group, which was clustered with I. exustus from India (GenBank accession no. AY577492), the Philippines (GenBank accession no. HM756308), and Thailand (GenBank accession no. AY282598), with the highest (99%) bootstrap support values for each ML and NJ method (Fig. 2).

Maximum likelihood phylogenetic tree of I. exustus based on a partial 18S rDNA (339 bp) (A) and 28S rDNA (1036 bp) (B) sequences. The bootstrap values for ML (left) and NJ (right) ≥50% are represented at each node of the phylogenetic tree. The scale bar represents 5 nucleotide substitutions for every 1000 nucleotides for the 18S rRNA gene and 2 nucleotide substitutions for every 100 nucleotides for the 28S rRNA gene. Bold letters indicate the sequences generated in the present study. Radix auricularia and Physa acuta were used as the outgroups
The phylogenetic analysis based on the 28S rDNA sequences (1036 bp) for the 45 I. exustus samples (44 sequences from the present study and one from GenBank) revealed one group, which was clustered with only I. exustus from Thailand (GenBank accession no. AF435662), with branch support values of 99% for each ML and NJ method (Fig. 2). These results showed that there is only one group in the genetic structure of I. exustus in Thailand, based on the 18S and 28S rDNA sequence analysis.
Our results for the phylogenetic group are inconsistent with those from previous studies reported for other genes (Gauffre-Autelin et al. 2017; Liu et al. 2010; Mouahid et al. 2018; Saijuntha et al. 2021). Previous phylogenetic studies based on mitochondrial sequences revealed the presence of three (Liu et al. 2010), four (Devkota et al. 2015), and five (Gauffre-Autelin et al. 2017; Saijuntha et al. 2021) clades of I. exustus. Thus, the present study indicated the low genetic diversity of I. exustus based on 18S and 28S rDNA sequence analysis. However, our results are in concordance with those of a previous study showing a Thai I. exustus strain clustering with those from Nepal, Oman, Sri Lanka, Indonesia, Malaysia, Myanmar, and the Philippines (Saijuntha et al. 2021).
Genetic diversity and haplotype network analyses
The 18S (339 bp) and 28S rDNA (1036 bp) sequences from the 44 I. exustus samples, which represented 21 provinces from Thailand, were genetically analyzed. The 28S rDNA gene of I. exustus displayed the highest intraspecific genetic divergence, which ranged from 0 to 0.78%. On the other hand, no variation was observed in the 18S rDNA gene, whose pairwise genetic distance was 0%, indicating the presence of a single haplotype.
Median-joining haplotype network analysis based on 28S rDNA from 44 I. exustus sequences revealed the presence of 8 different haplotypes (H1–H8) with 17 variable sites (Table 3). The genetic distances among the haplotypes varied, ranging from 0.001 to 0.010 (Table 4). Of these, 6 haplotypes (H2, H3, H4, H6, H7, and H8) were unique, and 2 haplotypes (H1 and H5) were shared by at least two regions. Haplotype H1 was the most widely distributed, covering all regions of Thailand included in this study, and accounting for 81.82% of all samples and 100% of all populations. In contrast, haplotype H5 was mainly distributed in Central and Southern Thailand, accounting for 4.54% of all samples and 33.33% of all populations. Thus, haplotype H1 was considered the major haplotype. The remaining haplotypes were found to be unique to a particular region, such as haplotype H2, which was found only in the northern region; H3 and H4, which were unique to the south; H6 and H7, which were present in the northeastern region; and H8, which was found only in the central region of Thailand (Fig. 3, Table 5). The genetic divergence in each population ranged from 0% in the populations from the western and eastern regions to 0.24% in the population from the northern region, with an average of 0.09% (Table 5).

Median-joining haplotype networks of I. exustus samples from various regions in Thailand based on 28S rDNA sequences. The geographical distributions of haplotypes are indicated by different colors, with circle sizes reflecting haplotype frequencies. Median vectors (small black dots) represent ancestral haplotypes that were either not sampled or missing
In our study, the I. exustus 28S rDNA sequences showed intraspecific genetic divergence ranging from 0 to 0.78% and were classified into 8 different haplotypes. In contrast, the 18S rDNA data showed no variation as revealed by a single haplotype among all sampled individuals. This suggested that the 18S rDNA gene had a relatively slow evolutionary rate and low variability compared to those of the 28S rDNA gene (Bargues and Mas-Coma 1997; Matsuda et al. 2014; Zein-Eddine et al. 2014). These results were consistent with those of a previous study on the genetic variation of the 18S and 28S rDNA genes of a freshwater snail in the genus Bulinus, which is a sister group of I. exustus (Jørgensen et al. 2011; Zein-Eddine et al. 2014). Comparatively, when examining genetic divergence among the genetic markers in I. exustus in previous reports, the intraspecific genetic distance values of the COI, 16S rDNA, ITS1, and ITS2 sequences were 0–5.33%, 0–2.34%, 0–2.21%, and 0–1.57%, respectively (Mouahid et al. 2018). These results indicated that the 28S rDNA had intraspecific genetic distance values that were closer to those of the nuclear ITS2 region than those of the nuclear ITS1 region and the mitochondrial COI and 16S rDNA genes. Generally, mitochondrial genes evolve faster than nuclear rDNA genes, accumulating a higher degree of sequence variation. This high intraspecies conservation makes nuclear rDNA genes helpful markers for resolving higher taxonomic levels or for use in biodiversity studies (Choudhary et al. 2015; Hwang and Kim 1999; Patwardhan et al. 2014; Pawlowski et al. 2012). Our findings showed that nuclear 18S and 28S rDNA had lower intraspecies discrimination power than mitochondrial COI and 16S rDNA markers (Matumba et al. 2020).
Haplotype network analysis based on 28S rDNA sequences of I. exustus from Thailand revealed 8 different haplotypes. Six haplotypes were unique, and two haplotypes were shared by at least two regions. Haplotype H1 was the major haplotype and was widely distributed across all regions of Thailand. However, the number of 28S rDNA haplotypes in the present study was lower than the number of mitochondrial sequence haplotypes in a previous report. Saijuntha et al. (2021) reported high genetic diversity with 21 haplotypes of I. exustus in Thailand based on the COI sequence. In addition, a previous study (Saijuntha et al. 2021) also found that I. exustus in several regions of Thailand shared haplotypes and that some regions had unique haplotypes, which was congruent with our results. The shared haplotypes in several regions may be due to the gene flow between I. exustus populations in Thailand (Saijuntha et al. 2021). The possibility of gene flow may be associated with water flow, the migration of waterbirds, and human activities (Bunchom et al. 2021; Habib et al. 2021; Neubauer et al. 2017; van Leeuwen and van der Velde 2012). The high stream velocities in canals or rivers might cause snails to be carried by the water current, resulting in a rise in gene flow from upstream to downstream populations (Bunchom et al. 2021; Habib et al. 2021). Simultaneously, the migration of waterbirds results in snails dispersing over large distances because the snails can become attached to waterbird bodies and feathers (Neubauer et al. 2017; van Leeuwen and van der Velde 2012). Furthermore, the occurrence of I. exustus in areas distant from its “native” geographic range may be associated with human activities of trading aquatic plants to which I. exustus can attach. This scenario stands as a key factor contributing to the migration and gene flow of I. exustus both in Thailand and globally (Pointier et al. 2005). These phenomena lead to the genetic homogenization of snail populations over long distances (Gow et al. 2007; Habib et al. 2021; Zein-Eddine et al. 2017). In contrast, the genetic differentiation in I. exustus may result from bottlenecks and genetic drift (Maes et al. 2022; Nyström et al. 2006). In addition, seasonal extinction events and a high rate of self-fertilization in populations may also explain the levels of genetic differences in I. exustus (Maes et al. 2022; Ryland and Bishop 1990). These phenomena commonly occur in the Planorbidae (Maes et al. 2022; Saijuntha et al. 2021). The distinct rivers in various regions of Thailand could impact the genetic variations and structure of I. exustus due to the water flow being limited or non-existent water flow between different catchments. This limitation in water flow might consequently restrict gene flow and migration among I. exustus populations across catchments, resulting in high genetic differences between areas (Bunchom et al. 2021). Meanwhile, changes in the natural environment, particularly seasonal changes, play a significant role. Seasonal variations in the availability of aquatic habitats are another factor affecting the fluctuations of snails. Adult I. exustus snails exhibit high desiccation tolerance, enabling them to survive the dry season by burying themselves in mud, whereas the resistance of juvenile snails is very low (Parashar and Rao 1982). Populations that survive a bottleneck may experience genetic drift (Nyström et al. 2006). Population bottlenecks lead to a reduction in population size and loss of genetic variation due to random genetic drift, resulting in diminished genetic variability within the population (Shirk et al. 2014; Weber et al. 2004).
The dispersal of snails into new areas may lead to a relationship between host and parasite and to the development of specific life cycles (Lv et al. 2009). The presence of snail intermediate hosts affects the distribution of trematode parasites (Dung et al. 2013; Maes et al. 2022). In the past, there were reports that I. exustus acts as the first intermediate host for several trematode parasites, especially echinostomes and schistosomes (Devkota et al. 2015; Krailas et al. 2022; Wiroonpan et al. 2021). The invasion of snails leads to parasites spreading faster and increases the chances of them infecting definitive hosts, including humans (Lu et al. 2018). In addition, the wide diffusion of snails due to natural factors results in pathogens spreading into new areas because parasites are likely to be translocated with their hosts (Demiaszkiewicz 2014; Kelehear et al. 2013; Taraschewski 2006).
In summary, we have assessed the suitability of two genetic markers for the molecular systematics and molecular identification of I. exustus. The nuclear 18S and 28S rDNA genes were successfully employed to distinguish I. exustus from another genus belonging to the family Planorbidae. We also revealed that the 18S rDNA gene had a relatively slow evolutionary rate and low variability compared to those of the 28S rDNA gene. The 28S rDNA gene of I. exustus is suitable for phylogenetic studies, whereas the 18S rDNA gene is appropriate for identification. However, these markers, particularly the 18S, may not be suitable for genetic analysis within the species, particularly for population genetic studies, due to their limited variation as seen in this study. The 18S and 28S rDNA genes may be useful as alternative, good genetic markers for identifying species of I. exustus.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
References
Andrus PS, Stothard JR, Kabatereine NB, Wade CM (2023) Comparing shell size and shape with canonical variate analysis of sympatric Biomphalaria species within Lake Albert and Lake Victoria, Uganda. Zool J Linn Soc:zlad052. https://doi.org/10.1093/zoolinnean/zlad052
Ardpairin J, Dumidae A, Subkrasae C, Nateeworanart S, Thanwisai A, Vitta A (2022) Preliminary survey of larval trematodes in freshwater snails of Phitsanulok Province in lower Northern Thailand. Iran J Parasitol 17:268–276. https://doi.org/10.18502/ijpa.v17i2.9545
AVMA (American Veterinary Medical Association) (2020) AVMA guidelines for the euthanasia of animals: 2020 editions. Available online: https://www.avma.org/KB/Policies/Documents/euthanasia.pdf. Accessed 20 Jul 2020
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16(1):37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Bargues MD, Mas-Coma S (1997) Phylogenetic analysis of Lymnaeid snails based on 18S rDNA sequences. Mol Biol Evol 14(5):569–577. https://doi.org/10.1093/oxfordjournals.molbev.a025794
Bawm S, Khaing NHE, Win SY, Thein SS, Khaing Y, Thaw YN, Soe NC, Chel HM, Hmoon MM, Hayashi N, Htun LL, Katakura K, Nonaka N, Nakao R (2022) Morphological and molecular identification of trematode cercariae related with humans and animal health in freshwater snails from a lake and a dam in Myanmar. Parasitol Res 121(2):653–665. https://doi.org/10.1007/s00436-022-07428-4
Brandt RAM (1974) The non-marine aquatic Mollusca of Thailand. Arch Molluskenkd 105:1–423
Bunchom N, Saijuntha W, Vaisusuk K, Pilap W, Suksavate W, Suganuma N, Agatsuma T, Petney TN, Tantrawatpan C (2021) Genetic variation of a freshwater snail Hydrobioides nassa (Gastropoda: Bithyniidae) in Thailand examined by mitochondrial DNA sequences. Hydrobiologia 848:2965–2976. https://doi.org/10.1007/s10750-019-04013-2
Chai JY (2019) Human intestinal flukes from discovery to treatment and control. Springer Nature B.V, Dordrecht, Netherlands
Choudhary K, Verma AK, Swaroop S, Agrawal N (2015) A review on the molecular characterization of digenean parasites using molecular markers with special reference to ITS region. Helminthologia 52:167–187. https://doi.org/10.1515/helmin-2015-0031
Cowie RH, Dillon RT, Robinson DG, Smith JW (2009) Alien non-marine snails and slugs of priority quarantine importance in the United States: a preliminary risk assessment. Am Malacol Bull 27:113–132. https://doi.org/10.4003/006.027.0210
Demiaszkiewicz AW (2014) Migrations and the introduction of wild ruminants as a source of parasite exchange and emergence of new parasitoses. Ann Parasitol 60(1):25–30
Devkota R, Brant SV, Loker ES (2015) The Schistosoma indicum species group in Nepal: presence of a new lineage of schistosome and use of the Indoplanorbis exustus species complex of snail hosts. Int J Parasitol 45(13):857–870. https://doi.org/10.1016/j.ijpara.2015.07.008
Dumidae A, Janthu P, Subkrasae C, Polseela R, Mangkit B, Thanwisai A, Vitta A (2021) Population genetics analysis of a Pomacea snail (Gastropoda: Ampullariidae) in Thailand and its low infection by Angiostrongylus cantonensis. Zool Stud 60:31. https://doi.org/10.6620/ZS.2021.60-31
Dung BT, Doanh PN, The DT, Loan HT, Losson B, Caron Y (2013) Morphological and molecular characterization of lymnaeid snails and their potential role in transmission of Fasciola spp. in Vietnam. Korean J Parasitol 51:657–662. https://doi.org/10.3347/kjp.2013.51.6.657
Frandsen F (1983) A field guide to freshwater snails in countries of the WHO Eastern Mediterranean region. Danish Bilharziasis Laboratory, Copenhagen, Denmark
Gauffre-Autelin P, von Rintelen T, Stelbrink B, Albrecht C (2017) Recent range expansion of an intermediate host for animal schistosome parasites in the indo-Australian Archipelago: phylogeography of the freshwater gastropod Indoplanorbis exustus in south and Southeast Asia. Parasit Vectors 10(1):126. https://doi.org/10.1186/s13071-017-2043-6
Gow JL, Noble LR, Rollinson D, Tchuenté LAT, Jones CS (2007) Contrasting temporal dynamics and spatial patterns of population genetic structure correlate with differences in demography and habitat between two closely related African freshwater snails. Biol J Linn Soc 90:747–760. https://doi.org/10.1111/j.1095-8312.2007.00771.x
Habib MR, Lv S, Rollinson D, Zhou X (2021) Invasion and dispersal of Biomphalaria species: increased vigilance needed to prevent the introduction and spread of schistosomiasis. Front Med 8:614797. https://doi.org/10.3389/fmed.2021.614797
Hwang UW, Kim W (1999) General properties and phylogenetic utilities of nuclear ribosomal DNA and mitochondrial DNA commonly used in molecular systematics. Korean J Parasitol 37(4):215–228. https://doi.org/10.3347/kjp.1999.37.4.215
Ikeda T, Iwasaki K, Suzuki T, Wong LJ, Pagad S (2021) Global register of Introduced and Invasive Species- Japan. Version 1.2. Invasive Species Specialist Group ISSG. Checklist dataset. https://doi.org/10.15468/nt2yla
Jørgensen A, Madsen H, Nalugwa A, Nyakaana S, Rollinson D, Stothard JR, Kristensen TK (2011) A molecular phylogenetic analysis of Bulinus (Gastropoda: Planorbidae) with conserved nuclear genes. Zool Scr 40(2):126–136. https://doi.org/10.1111/j.1463-6409.2010.00458.x
Kelehear C, Brown GP, Shine R (2013) Invasive parasites in multiple invasive hosts: the arrival of a new host revives a stalled prior parasite invasion. Oikos 122:1317–1324. https://doi.org/10.1111/j.1600-0706.2013.00292.x
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120. https://doi.org/10.1007/BF01731581
Krailas D, Namchote S, Komsuwan J, Wongpim T, Apiraksena K, Glaubrecht M, Sonthiporn P, Sansawang C, Suwanrit S (2022) Cercarial dermatitis outbreak caused by ruminant parasite with intermediate snail host: schistosome in Chana. South Thailand Evol Syst 6(2):151–173. https://doi.org/10.3897/evolsyst.6.87670
Kristensen TK, Ogunnowo O (1987) Indoplanorbis exustus (Deshaies, 1834), a freshwater snail new for Africa, found in Nigeria (Pulmonata: Planorbidae). J Molluscan Stud 53:245–246. https://doi.org/10.1093/mollus/53.2.245
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Leigh JW, Bryant D (2015) Popart: full-feature software for haplotype network construction. Methods Ecol Evol 6(9):1110–1116. https://doi.org/10.1111/2041-210X.12410
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Liu L, Mondal MM, Idris MA, Lokman HS, Rajapakse PJ, Satrija F, Diaz JL, Upatham ES, Attwood SW (2010) The phylogeography of Indoplanorbis exustus (Gastropoda: Planorbidae) in Asia. Parasit Vectors 3:57. https://doi.org/10.1186/1756-3305-3-57
Lu XT, Gu QY, Limpanont Y, Song LG, Wu ZD, Okanurak K, Lv ZY (2018) Snail-borne parasitic diseases: an update on global epidemiological distribution, transmission interruption and control methods. Infect Dis Poverty 7:28. https://doi.org/10.1186/s40249-018-0414-7
Lv S, Zhang Y, Liu HX, Hu L, Yang K, Steinmann P, Chen Z, Wang LY, Utzinger J, Zhou XN (2009) Invasive snails and an emerging infectious disease: results from the first national survey on Angiostrongylus cantonensis in China. PLoS Negl Trop Dis 3:e368. https://doi.org/10.1371/journal.pntd.0000368
Maes T, De Corte Z, Vangestel C, Virgilio M, Smitz N, Djuikwo-Teukeng FF, Papadaki MI, Huyse T (2022) Large-scale and small-scale population genetic structure of the medically important gastropod species Bulinus truncatus (Gastropoda, Heterobranchia). Parasit Vectors 15:328. https://doi.org/10.1186/s13071-022-05445-x
Matsuda T, Morishita M, Hinomoto N, Gotoh T (2014) Phylogenetic analysis of the spider mite sub-family Tetranychinae (Acari: Tetranychidae) based on the mitochondrial COI gene and the 18S and the 59 end of the 28S rRNA genes indicates that several genera are polyphyletic. PLoS One 9(10):e108672. https://doi.org/10.1371/journal.pone.0108672
Matumba TG, Oliver J, Barker NP, McQuaid CD, Teske PR (2020) Intraspecific mitochondrial gene variation can be as low as that of nuclear rRNA. F1000Res 9:339. https://doi.org/10.12688/f1000research.23635.2
Meyer JH, Fourdrigniez M, Soubeyran Y, Pagad S (2021) Global Register of Introduced and Invasive Species-French Polynesia. Version 1.2. Invasive Species Specialist Group ISSG. Checklist dataset. https://doi.org/10.15468/bi2dvk
Morgan JA, DeJong RJ, Jung Y, Khallaayoune K, Kock S, Mkoji GM, Loker ES (2002) A phylogeny of planorbid snails, with implications for the evolution of Schistosoma parasites. Mol Phylogenet Evol 25(3):477–488. https://doi.org/10.1016/s1055-7903(02)00280-4
Mouahid G, Clerissi C, Allienne JF, Chaparro C, Al Yafae S, Mintsa Nguema R, Ibikounlé M, Moné H (2018) The phylogeny of the genus Indoplanorbis (Gastropoda, Planorbidae) from Africa and the French West Indies. Zool Scr 47:558–564. https://doi.org/10.1111/zsc.12297
Mouchet F, Rey JL, Cunin P (1987) Decouverte d’Indoplanorbis exustus (Planorbidae; Bulininae) a Yamoussokro, Cote d’Ivoire. Bull Soc Pathol Exot Filiales 80:811–812
Neubauer TA, Mandic O, Harzhauser M, Jovanović G (2017) The discovery of Bulinus (Pulmonata: Planorbidae) in a Miocene palaeolake in the Balkan Peninsula. J Molluscan Stud 83(3):295–303. https://doi.org/10.1093/mollus/eyx015
Nithiuthai S, Anantaphruti MT, Waikagul J, Gajadhar A (2004) Waterborne zoonotic helminthiases. Vet Parasitol 126:167–193. https://doi.org/10.1016/j.vetpar.2004.09.018
Nyström V, Angerbjörn A, Dalén L (2006) Genetic consequences of a demographic bottleneck in the Scandinavian arctic fox. Oikos 114:84–94. https://doi.org/10.1111/j.2006.0030-1299.14701.x
Pagad S (2020) Protected areas - Global Register of Introduced and Invasive Species - Socotra Archipelago, Yemen. Invasive Species Specialist Group ISSG. Checklist dataset. https://doi.org/10.15468/n9mssm
Parashar BD, Rao KM (1982) Effect of desiccation on freshwater snail, Indoplanorbis exustus, intermediate host of schistosomiasis. Jpn J Med Sci Biol 35:243–247. https://doi.org/10.7883/yoken1952.35.243
Patwardhan A, Ray S, Roy A (2014) Molecular markers in phylogenetic studies-a review. J Phylogen Evolution Biol 2:2. https://doi.org/10.4172/2329-9002.1000131
Pawlowski J, Audic S, Adl S, Bass D, Belbahri L, Berney C, Bowser SS, Cepicka I, Decelle J, Dunthorn M, Fiore-Donno AM, Gile GH, Holzmann M, Jahn R, Jirků M, Keeling PJ, Kostka M, Kudryavtsev A, Lara E et al (2012) CBOL Protist Working Group: barcoding eukaryotic richness beyond the animal, plant, and fungal Kingdoms. PLoS Biol 10(11):e1001419. https://doi.org/10.1371/journal.pbio.1001419
Pointier JP, David P, Jarne P (2005) Biological invasions: the case of planorbid snails. J Helminthol 79:249–256
Ryland JS, Bishop JDD (1990) Prevalence of cross-fertilisation in the hermaphroditic compound ascidian Diplosoma listerianum. Mar Ecol Prog Ser 61:125–132. https://doi.org/10.3354/meps061125
Saijuntha W, Tantrawatpan C, Agatsuma T, Rajapakse R, Karunathilake KJK, Pilap W, Tawong W, Petney TN, Andrews RH (2021) Phylogeographic genetic variation of Indoplanorbis exustus (Deshayes, 1834) (Gastropoda: Planorbidae) in South and Southeast Asia. One Health 12:100211. https://doi.org/10.1016/j.onehlt.2021.100211
Shirk RY, Hamrick JL, Zhang C, Qiang S (2014) Patterns of genetic diversity reveal multiple introductions and recurrent founder effects during range expansion in invasive populations of Geranium carolinianum (Geraniaceae). Heredity 112:497–507. https://doi.org/10.1038/hdy.2013.132
Sil M, Mahveen J, Roy A, Karanth KP, Aravind NA (2022) Insight into the evolutionary history of Indoplanorbis exustus (Bulinidae: Gastropoda) at the scale of population and species. Biol J Linn Soc Lond 137(1):68–84. https://doi.org/10.1093/biolinnean/blac062
Stothard JR, Bremond P, Andriamaro L, Loxton NJ, Sellin B, Sellin E, Rollinson D (2000) Molecular characterization of the freshwater snail Lymnaea natalensis (Gastropoda: Lymnaeidae) on Madagascar with an observation of an unusual polymorphism in ribosomal small subunit genes. J Zool 252:303–315. https://doi.org/10.1111/j.1469-7998.2000.tb00625.x
Tamura K (1992) Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. 9(4):678-687. https://doi.org/10.1093/oxfordjournals.molbev.a040752
Taraschewski H (2006) Hosts and parasites as aliens. J Helminthol 80:99–128. https://doi.org/10.1079/joh2006364
Van Bocxlaer B, Strong EE, Richter R, Stelbrink B, Von Rintelen T (2018) Anatomical and genetic data reveal that Rivularia Heude, 1890 belongs to Viviparinae (Gastropoda: Viviparidae). Zool J Linnean Soc 182(1):1–23. https://doi.org/10.1093/zoolinnean/zlx014
van Leeuwen CHA, van der Velde G (2012) Prerequisites for flying snails: external transport potential of aquatic snails by waterbirds. Freshw Sci 31:963–972. https://doi.org/10.1899/12-023.1
Vences M, Thomas M, Bonett RM, Chiari Y, Vietes DR (2005) Deciphering amphibian diversity through DNA barcoding: chances and challenges. Philos Trans R Soc Lond Ser B Biol Sci 360(1462):1859–1868. https://doi.org/10.1098/rstb.2005.1717
Weber DS, Stewart BS, Lehman N (2004) Genetic consequences of a severe population bottleneck in the Guadalupe fur seal (Arctocephalus townsendi). J Hered 95:144–153. https://doi.org/10.1093/jhered/esh018
Wiroonpan P, Chontananarth T, Purivirojkul W (2021) Cercarial trematodes in freshwater snails from Bangkok, Thailand: prevalence, morphological and molecular studies and human parasite perspective. Parasitology 148(3):366–383. https://doi.org/10.1017/S0031182020002073
Won EJ, Lee YJ, Kim MJ, Chai JY, Na BK, Sohn WM (2020) Morphological and molecular characteristics of Clinostomid Metacercariae from Korea and Myanmar. Korean J Parasitol 58(6):635–645. https://doi.org/10.3347/kjp.2020.58.6.635
Zein-Eddine R, Djuikwo-Teukeng FF, Al-Jawhari M, Senghor B, Huyse T, Dreyfuss G (2014) Phylogeny of seven Bulinus species originating from endemic areas in three African countries, in relation to the human blood fluke Schistosoma haematobium. BMC Evol Biol 14:271. https://doi.org/10.1186/s12862-014-0271-3
Zein-Eddine R, Djuikwo-Teukeng FF, Dar Y, Dreyfuss G, Van den Broeck F (2017) Population genetics of the Schistosoma snail host Bulinus truncatus in Egypt. Acta Trop 172:36–43. https://doi.org/10.1016/j.actatropica.2017.04.002
Acknowledgements
We would like to thank the Department of Microbiology and Parasitology, Faculty of Medical Science, Naresuan University, for providing the necessary support and access to facilities.
Funding
This study was supported by Naresuan University (NU) and the National Science, Research and Innovation Fund (NSRF), Thailand (grant no. R2566B046). This funding sponsor had no role in the design of the study.
Author information
Authors and Affiliations
Contributions
AD performed experiments, analyzed data, and wrote the paper. CS performed experiments and wrote the paper. JA performed experiments and wrote the paper. SP performed experiments and wrote the paper. CH performed experiments and wrote the paper. AT provided materials and wrote the paper. AV performed experiments, analyzed data, provided materials, and wrote the paper.
Corresponding author
Ethics declarations
Ethical approval
The experiments involving invertebrate animals (snails) were approved from the Center for Animal Research at Naresuan University (Project Ethics No: NU-AQ640803).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Julia Walochnik
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dumidae, A., Subkrasae, C., Ardpairin, J. et al. Genetic variation of the freshwater snail Indoplanorbis exustus (Gastropoda: Planorbidae) in Thailand, inferred from 18S and 28S rDNA sequences. Parasitol Res 123, 93 (2024). https://doi.org/10.1007/s00436-024-08120-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00436-024-08120-5