
Abstract
Depth profiles of Fe, Mn, (HS)t, Cu and Cd concentrations in pore water were determined on a seasonal scale in intertidal sediments of Ria Formosa. Concentrations of Cu and Cd were also determined in near-bottom water during the short period that water inundates the sediment. A maximum near the sediment-water interface was observed in depth profiles of Mn and Fe concentrations followed by a decrease with depth. Otherwise, depth profiles of (HS−)t were irregular but peak concentrations was observed below Mn and Fe maximum. Although subsurface maximum was observed at deeper layers for Cu and Cd, the profiles shape varied among sites and sampling dates. This suggests site specificity and alterations associated with early diagenetic reactions. In order to assess exchanges of Cu and Cd across the sediment water interface, diffusive fluxes and advective transport were estimated. Both contribute substantially to the daily transfer of Cd from intertidal sediments to the water column of Ria Formosa. In the case of Cu, the flux associated with tidal flooding (advective flux) was the major contributor. Presumably, the exchange of trace elements between the sediment-water interface in intertidal areas of macro- and meso-tidal systems are underestimated since do not take into consideration the pulse contribution associated with tidal flooding.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recent sediments of estuarine systems comprise the first few centimetres below the sediment-water interface. This layer contains organic matter recently imported from overlying water or produced within the sediment. The degradation of organic matter in undisturbed sediments reflects a well-established depth zonation of redox reactions: oxygen reduction near the sediment-water interface; reduction of nitrate, manganese oxides, iron oxides and sulphate (Dhakar & Burdige, 1996; Aller, 1998). The thickness of each zone depends on the changes of productivity, oxygen concentration in near-bottom water, shifting in relative positions of reducing and oxidizing sediment components and the population of microorganisms (Aller, 1994; Anschutz et al., 2000; Hyacinthe et al., 2001). As a result, early diagenetic reactions lead both to changes in pore water composition and to generation of new mineral phases (Berner, 1980; van Cappellen & Wang, 1996). It is generally recognized that geochemistry of trace elements are closely linked to the diagenetic reactions and to the interaction with forms of Fe, Mn and S (Shaw et al., 1990; Thomson et al., 2001; Fones et al., 2004; Naylor et al., 2004).
Permeable intertidal sediments are a major feature macro- and mesotidal ecosystems. In these areas, the tide alternately inundates and exposes the sediment to the atmosphere. This covering and uncovering situations create non-steady state conditions between solids and pore water. When sediments are covered with tidal water, diffusion is the main transport mechanism across the sediment-water interface. During the flooding period, advective transport induces chemical reactions and mass fluxes through the sediment-water interface influencing the ecology of the marine environment (Hemond et al., 1984; Huettel et al., 1998). Several changes associated with this physical process have been reported in the literature: supply of oxygen and nutrients to deeper sediment layers (Kerner & Wallman, 1992; Huettel et al., 1998); ammonium export to the water column (Falcão & Vale, 1995; Rocha, 1997); phosphate removal to the solid fraction (Falcão & Vale, 1990); and oxidation of pore water Mn(II) and Fe(II) in minute time scale (Caetano et al., 1997). Most of these studies have focused on nutrient regeneration and less attention was dedicated to trace metals in pore waters and flooding waters, although this dynamic may have major importance to benthic organisms.
Ria Formosa is a coastal lagoon located in the south coast of Iberian Peninsula. This lagoon may be considered an ideal field laboratory to this study since the intertidal area reaches 2/3 of the wet lagoon area (7000 ha). The lagoon has no freshwater inputs and 50–75% of water mass exchanges each semi diurnal tidal cycle. Low levels of Cu (3.8 ± 0.6 nM) and Cd (0.14 ± 0.08 nM) in coastal waters adjacent to the lagoon suggest minor export of these metals from Ria Formosa (Caetano & Vale, 2003). However, trace metal concentrations in muddy sediments contrast with the background levels found in sandy sediments from the main channels (Caetano et al., 2002; Bebianno, 1995) and Bebianno & Serafim (2003) measured high levels of metallothionein induced by Cd contamination in natural populations of clams [Ruditapes decussatus (Linneus, 1758)]. Due to intense production (300 tonnes y−1) of Ruditapes decussatus and Venerupis pullastra (Montagu, 1803) in intertidal sediments a major concern has arisen concerning the environmental quality required to maintain sustainable clams production.
This study examines the seasonal variation of Cu and Cd concentrations in the upper intertidal sediments of Ria Formosa. Simultaneous determinations of Fe, Mn and sulphide concentrations were used to obtain a better understanding of Cu and Cd exchanges between pore waters and solids. Diffusive and advective fluxes of Cu and Cd across the sediment-water interface of intertidal sediments were calculated.
Sampling and methods
Sampling
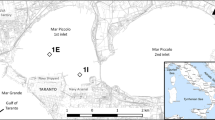
Short sediments cores (4 cm depth) were collected around low tide in two stations (A and G) of Ria Formosa intertidal area every three months from July 1992 to March 1993 (Fig. 1). Sediment cores were sliced in loco in 0.5 cm layers, stored in acid precleaned HDPE vials avoiding air presence inside and kept in refrigerated atmosphere (Caetano et al., 1995). In the laboratory, pore water was separated by centrifugation (20 min; 3000 rpm) and immediately filtered through 0.45 μm polycarbonate membranes. Filtration and subsequent manipulation of the samples were carried out in a glove box under N2 atmosphere. Pore water samples were divided in two sub-samples: one for total hydrogen sulphide (HS−)t analysis, and the other acidified with suprapure HNO3 (pH < 2) to determine total concentrations of Fe, Mn, Cu and Cd. At station A, flooding waters were collected 1 cm above the sediment surface directly into pre-cleaned syringes over the short interval of time that water floods the sediments. Flooding water samples were immediately filtered and acidified for Cu and Cd analysis.

Map of Ria Formosa with the sampling locations, station A and G
Pore water analysis
Total dissolved Mn and Fe in pore waters were determined by atomic absorption spectrometry using direct aspiration into air-acetylene flame within 2 days after sampling. Concentrations were determined with the standard additions method. Detection limits and reproducibility were 0.3 μM and 5% for Mn and 1.0 μM and 2.3% for Fe. Recovery of standard solutions of Fe and Mn ranged from 92 to 98%. Levels of (HS−)t in pore waters were determined by differential pulse cathodic striping voltammetry or differential pulse polarography, depending on the sulphide concentration (Luther et al., 1985; Luther & Tsamakis, 1989). Dissolved sulphide, as the joint response of H2S, HS− and S2−, was analysed by taking aliquots of 0.5–1.0 mL, added to 5 mL of a NaCl solution at pH 10–12 with NaOH, which had been purged with N2 for 4 min to remove oxygen. Sulphide was determined on the hanging mercury drop electrode and on the static dropping mercury electrode using an Ag/ AgCl / NaCl reference system and a platinum electrode as auxiliary. Recovery of standard sulphide solutions was 97%. Evaluation of the sulphur was done by constructing calibration curves for diluted samples and using standard addition method for undiluted samples. Total dissolved Cu and Cd were concentrated through complexing, with a buffered “mix solution” of ammonium pyrrolidine dithiocarbamate (APDC) and diethylammonium diethyldithiocarbamate (DDDC) and extracted into chloroform. The procedure was completed by a back extraction into nitric acid (Danielsson et al., 1978). This method was adapted to samples with small volumes (4–10 mL). We used 5 mL of chloroform, 250 μL of “mix solution” and 100 μL of buffer solution for a 10 mL sample. The reagents used were suprapure grade or purified. The modified methodology was tested with 12 sub-samples of two certified reference materials (NASS-3, CASS-3). Trace metal concentrations obtained (Table 1) were not statistically different from their certified ones (t-student; α = 0.05). Copper and Cd were measured with a Perkin-Elmer transversely heated graphite furnace atomic absorption spectrometer with Zeeman background correction and concentrations determined with the standard additions method. The detection limit for Cu was 1.4 nM and for Cd 0.06 nM and the reproducibility 1 and 10%, respectively.
Results and discussion
Iron, manganese and sulphur
The profiles of Fe, Mn and (HS−)t concentration in pore waters are presented in Fig. 2. Profiles of Mn concentrations were similar in the two studied stations consisting in a maximum near the sediment-water interface followed by a rapid decrease with depth. Iron profiles also showed a similar depth variation with a peak maximum at the same depths as Mn or just below. The sharp increase of both metal concentration in the 0.5–1 cm sediment layer indicates that penetration depth of O2 was restricted to the topmost sediments, as previous observed (Brotas et al., 1990). These results also indicate that the upper centimetre sediment layer is suboxic and dominated biogeochemically by Mn and Fe cycles. Dissolved Mn and Fe diffuses upward from the suboxic layers being retained closer to the sediment-water interface as authigenic Fe(III) oxides and Mn(IV) oxides and/or diffuses out of the sediment to the overlying water, as observed in classic studies (e.g. Aller, 1994). It is possible that variations in this sediment layer occur on a millimetre scale that could not be resolved by the sampling method employed here.

Depth profiles of Mn, Fe, (HS−)t concentrations (nM) in pore waters of stations A (■) and G (□)
In spite of the pronounced seasonal signal of primary production in the lagoon (Falcão & Vale, 1990) and consequently different inputs of organic matter to the sediments, both depth profiles of Mn and Fe concentrations showed no seasonal variation. This lack of variation suggests that fast recycling processes within the benthic layer mask the seasonal variations induced by changes of temperature or organic matter input (Caetano et al., 1995). In fact, Caetano et al. (1997) showed that the turnover period of Fe and Mn in intertidal sediments of Ria Formosa is lower than a semi-diurnal tidal cycle. The vertical distribution of (HS−)t concentration was scattered with high concentrations occurring close to low or undetectable concentrations. This scattering distribution suggests that sulphate reduction rates may differ between contiguous sediment layers. However, maximum (HS−)t concentrations were observed below Mn and Fe peaks, which is in accordance to the established depth sequence of diagenetic reactions governed by the preferential use of electron acceptors that yields the highest amount of free energy for the bacterially mediated oxidation of organic matter (Aller, 1998). As observed for Fe and Mn, sulphide concentration in pore waters showed no significant seasonal variation. This may be attributed to the fast recycling processes of Fe and Mn within the benthic layer that changes the reduction of sulphate during organic matter degradation.
Copper and cadmium
Depth profiles of Cu and Cd concentration in pore waters are shown in Fig. 3. In spite of the scattered depth variation, subsurface maximum were observed in several occasions. This scattering may result from the release of Cu and Cd associated with Fe and Mn oxides reduction. Several authors have proposed that trace metals are included/released in Fe and Mn oxides generated in upper sediment layers (Gobeil et al., 1987; Tessier et al., 1996). Thus, depth variation of pore water Cu and Cd concentrations at upper sediments may reflect the short turnover period (<12 h) of Fe and Mn redox reactions in Ria Formosa sediments (Caetano et al., 1997). Maximum concentrations at deeper layers reached higher values than those predicted by thermodynamic data based on concentrations of (HS−)t suggesting that the precipitation rate of authigenic Cu and Cd is not controlled by sulphide. Thus the high stability of organic or inorganic complexes of these metals (Davies-Colley et al., 1985; Gobeil et al., 1987) may influence the precipitation rate of CdS and CuS (Sundby et al., 2004). Furthermore, changes in the sulphide concentration due to different sulphate reduction rates may also influence the equilibrium between solid and pore water.

Depth profiles of Cu and Cd concentrations (nM) in pore waters of station A (■) and G (□)
Estimation of sediment-water exchanges
The high levels of Cu and Cd in pore waters of upper sediment layers than in overlying water imply their export to the water column. Since intertidal sediments are alternatively covered with water and exposed to the atmosphere, two types of fluxes are predicted: diffusive fluxes during the submerged period and advective transport associated with the tidal inundation. On the basis of the measured concentration profiles, the benthic fluxes of Cu and Cd, can be estimated by Fick’s first law of diffusion F dif = φ3 Ds (C0−Cp)/Δx (Berner, 1980), where φ is the porosity (0.75) calculated in Caetano (1998), Ds(Cu) = 6.25 x 10−6 cm2 s−1, Ds(Cd) = 6.23 x 10−6 cm2 s−1 (Li & Gregory, 1974), C0 = [Cu] or [Cd] in pore water of the topmost sediment layer, Cp = [Cu] or [Cd] in pore water and x is the space co-ordinate, negative into sediment and origin at sediment-water interface. The average diffusive fluxes of Cd were 10 ± 5 and 4 ± 4 nmol m−2 d−1 at stations G and A, respectively. The estimated fluxes of Cu were 257 ± 99 and 326 ± 259 nmol m−2 d−1. Studies performed by Falcão & Vale (1990) in Ria Formosa showed an advective transport of nutrients from pore water to the water column associated with the tidal inundation of permeable sediments. In order to investigate this advective transport of Cu and Cd, concentrations in flooding waters of station A were measured over the short inundation period (Fig. 4). In the first minutes of inundation levels of Cu in the flooding water were higher (130 nM) than concentrations in pore waters of the upper sediment layer (0–0.5 cm), and then decreased gradually to 16 nM in the subsequent 38 minutes of inundation. These temporal variation indicate that Cu was exported from sediment pore waters. Cadmium in the flooding water also decreased from 2.4 to 0.6 nM in the first 2 min increasing to 1.4 nM at the end of the inundation period. On the basis of these measurements one may calculate the advective transport (T) of these metals to the water column using the expression: T = Σ (Ct + 1−Ct) (ht + 1−ht)/ 2, where Ct + 1 and Ct are [Cu] or [Cd] in the flooding water at times t + 1 and t and ht + 1 and ht are the water depth at the same times. It was observed that water depth during the field measurements increased on the average 5 cm each 5 min of inundation. The transport of Cu and Cd was calculated for the first 38 min of inundation. Since intertidal sediments are inundated twice a day, the advective daily flux (T) was multiplied by a factor of two. The flux of Cd was 36 nmol m−2 d−1, while for Cu reached 16x103 nmol m−2 d−1.

Time course evolution of Cu (●) and Cd (▴) concentrations (nM) in flooding water during a 60 min tidal inundation period
Comparison of diffusive and advective fluxes
On a daily basis, the advective flux of Cu was two orders of magnitude higher than the diffusive flux, reinforcing the importance of pulse mechanisms associated with the tidal flushing on sediment-water exchanges. The gradual decrease of Cu concentration during the first 38 min of inundation (Fig. 4) suggests that Cu in pore waters remains in the solution presumably as organic complexes (Dai et al., 1995; Skrabal et al., 2000) and the efficiency of sorption or co-precipitation into Fe and Mn oxides is low. Advective and diffusive fluxes of Cd are of the same order of magnitude indicating similar significance of the two mechanisms on a daily scale. Advective export of Cd from sediments may be limited by sorption/co-precipitation into the freshly formed Fe oxides during the first minutes of tidal inundation (Caetano et al., 1997). Laboratory studies demonstrated that 80% of dissolved Cd is adsorbed on amorphous Fe2O3.H2O in a minute time scale (Benjamin & Leckie, 1981). Considering the magnitude and the periodicity of advective export, successive tidal inundation would deplete the pool of metals in the sediment if not balanced by equivalent inputs. Conservation of mass requires an import during the submerged period by settling of suspended particles, advection of water and deposition of particles during the ebb tide. In fact, an enrichment of dissolved Cu near the sediment-water interface was observed in the subsequent low tide suggesting that the kinetics involved in remobilization to pore waters were fast.
Diffusive fluxes of Cu and Cd in Ria Formosa sediments were comparable to values calculated for low contaminated ecosystems and much lower than polluted sites (Elbe River and Odiel River) (Table 2). However, taking into account the pulse advective flux, values may increase up to two orders of magnitude. These results emphasise the importance of the advective contribution to the budget of metals exchanged between permeable sediments and water in macro- and meso-tidal coastal ecosystems. One may speculate that sediment-water exchanges in most systems are often underestimated, because pulse mechanisms related to the tide may shift sediments from sink to source, and consequently their effect on organisms may be enhanced.
References
Aller, R., 1994. The sedimentary Mn cycle in long island sound: Its role as intermediate oxidant and the influence of bioturbation, O2 and Corg flux on diagenetic reaction balances. Journal of Marine Research 52: 259–295.
Aller, R., 1998. Mobile deltaic and continental shelf muds as suboxic, fluidized bed reactors. Marine Chemistry 61: 143–155.
Anschutz, P., B. Sundby, L. Lefrançois, G. Luther & A. Mucci, 2000. Interactions between metal oxides and species of nitrogen and iodine in bioturbated marine sediments. Geochimica et Cosmochimica Acta 64: 2751–2763.
Bebianno, M., 1995. Effects of pollutants in the Ria Formosa lagoon, Portugal. Science of the Total Environment 171: 107–115.
Bebianno, M. & A. Serafim, 2003. Variation of metal and metallothionein concentrations in a natural population of Ruditapes decussatus. Archives of Environmental Contamination and Toxicology 44: 53–66.
Benjamin, M. & J. Leckie, 1981. Multiple-site adsorption of Cd, Cu, Zn and Pb on amorphous iron oxyhydroxide. Journal of Colloid and Interface Science 79: 209-221.
Berner, R., 1980. Early diagenesis. A Theoretical Approach. Princeton University Press, Princeton, New Jersey.
Blasco, J., V. Sáenz & A. Gómez-Parra, 2000. Heavy metal fluxes at sediment–water interface of three coastal ecosystems from south-west of the Iberian Peninsula. Science of Total Environment 247: 189–199.
Brotas, V., A. Ferreira, C. Vale & F. Catarino, 1990. Oxygen profiles in intertidal sediments of Ria Formosa (S. Portugal). Hydrobiologia 207: 123–129.
Caetano, M., (1998). Biogeoquímica do manganês, ferro, cobre e cádmio em sedimentos da Ria Formosa. [PhD Thesis]. Universidade de Algarve, Faro.
Caetano, M., M. Falcão, C. Vale & M. Bebianno, 1997. Tidal flushing of ammonium, iron and manganese from inter-tidal sediment pore waters. Marine Chemistry 58: 203–211.
Caetano, M., M. Madureira, C. Vale, M. Bebianno & M. Gonçalves, 1995. Variations of Mn, Fe and S concentrations in sediment pore waters of Ria Formosa at different time scales. Netherlands Journal of Aquatic Ecology 29: 275–281.
Caetano, M. & C. Vale, 2003. Trace elemental composition of seston and plankton along the portuguese coast. Acta Oecologica 24: S341–S349.
Caetano, M., C. Vale & M. Bebianno, 2002. Distribution of Fe, Mn, Cu and Cd in upper sediments and sediment-trap material of Ria Formosa (Portugal). Journal of Coastal Research SI36: 118–123.
Cheevaporn, V., G. Jacinto & M. san Diego-McGlone, 1995. Heavy metals in Bang Pakong River Estuary, Thailand: sedimentary vs diffusive fluxes. Marine Pollution Bulletin 31: 290–294.
Ciceri, G., S. Maran, W. Martinotti & G. Queirazza, 1992. Geochemical cycling of heavy metals in a marine coastal area: benthic flux determination from pore water profiles and in situ measurements using benthic chambers. Hydrobiologia 235: 501–517.
Dai, M., J.-M. Martin & G. Cauwet, 1995. The significance role of colloids in the transport and transformation of organic carbon and associated metals (Cd, Cu and Ni) in the Rhone delta (France). Marine Chemistry 51: 159–175.
Danielsson, L., B. Magnusson & S. Westerlund, 1978. An improved metal extraction procedure for the determination of trace metals in sea water by atomic absorption spectrometry with electrotermal atomization. Analytica Chimica Acta 98: 47–57.
Davies-Colley, R., P. Nelson & K. Williamson, 1985. Sulfide control of cadmium and copper concentrations in anaerobic estuarine sediments. Marine Chemistry 16: 173–186.
Dhakar, S. & D. Burdige, 1996. A coupled, non-linear, steady state model for diagenetic processes in pelagic sediments. American Journal of Science 296: 296–330.
Falcão, M. & C. Vale, 1990. Study of the Ria Formosa ecosystem: benthic nutrient remineralization and tidal variability of nutrients in the water. Hydrobiologia 207: 137–146.
Falcão, M. & C. Vale, 1995. Tidal flushing of ammonium from intertidal sediments of Ria Formosa, Portugal. Netherlands Journal of Aquatic Ecology 29: 239–244.
Fones, G., W. Davison & J. Hamilton-Taylor, 2004. The fine scale remobilization of metals in the surface sediment of the North-East Atlantic. Continental Shelf Research 24: 1485–1504.
Gobeil, C., N. Silverberg, B. Sundby & D. Cossa, 1987. Cadmium diagenesis in Laurentian trough sediments. Geochimica et Cosmochimica Acta 51: 589–596.
Hemond, H., W. Nuttle, R. Burke & K. Stolzenbach, 1984. Surface infiltration in salt marshes: Theory, measurement, and biogeochemical implications. Water Resources Research 20: 591-600.
Huettel, M., W. Ziebis, S. Forster & G. Luther, 1998. Advective transport affecting metal and nutrient distributions and interfacial fluxes in permeable sediments. Geochimica et Cosmochimica Acta 62: 613–631.
Hyacinthe, C., P. Anschutz, P. Carbonel, J. Jouanneau & F. Jorinssen, 2001. Early diagenetic processes in muddy sediments of the Bay of Biscay. Marine Geology 177: 111-128.
Kerner, M. & K. Wallmann, 1992. Remobilization events involving Cd and Zn from intertidal flat sediments in the Elbe estuary during the tidal cycle. Estuarine, Coastal and Shelf Science 35: 371–393.
Li, Y. & S. Gregory, 1974. Diffusion of ions in sea water and in deep-sea sediments. Geochimica et Cosmochimica Acta 38: 703–714.
Luther, G., A. Giblin & R. Varsolona, 1985. Polarographic analysis of sulphur species in marine porewater. Limnology and Oceanography 30: 727–736.
Luther, G. & E. Tsamakis, 1989. Concentration and form of dissolved sulphide in the oxic water column of the ocean. Marine Chemistry 27: 165–177.
Naylor, C., W. Davison, M. Motelica-Heino, G. van Den Berg & L. van der Heijdt, 2004. Simultaneous release of sulfide with Fe, Mn, Ni and Zn in marine harbour sediment measured using metal/sulphide DGT probe. Science of the Total Environment 328: 275–286.
Petersen, W., K. Wallman, L. Pinglin, F. Schroeder & H. Knauth, 1995. Exchange of trace elements at the sediment–water interface during early diagenesis processes. Marine Freshwater Research 46: 19–26.
Rocha, C., 1997. Rhythmic ammonium regeneration and flushing in intertidal sediments of the Sado estuary. Limnology and Oceanography 43: 823–831.
Shaw, T., J. Gieskes & R. Jahnke, 1990. Early diagenesis in different depositional environments: The response of transition metals in pore water. Geochimica et Cosmochimica Acta 54: 1233–1246.
Skrabal, S., J. Donat & D. Burdige, 2000. Pore water distributions of dissolved copper and copper-complexing ligands in estuarine and coastal marine sediments. Geochimica et Cosmochimica Acta 64: 1843–1857.
Sundby, B., P. Martinez & C. Gobeil, 2004. Comparative geochemistry of cadmium, rhenium, uranium and molybdenum in continental margin sediments. Geochimica et Cosmochimica Acta 68: 2485–2494.
Tessier, A., D. Fortin, N. Belzile, R. De Vitre & G. Leppard, 1996. Metal sorption to diagenetic iron and manganese oxyhydroxides and associated organic matter: narrowing the gap between field and laboratory measurements. Geochimica et Cosmochimica Acta 60: 387–404.
Thomson, J., S. Nixon, I. Croudace, T. Pedersen, L. Brown, G. Cook & A. MacKenzie, 2001. Redox-sensitive element uptake in North-East Atlantic ocean sediments (benthic boundary layer experiment sites). Earth and Planetary Science Letters 184: 535–547.
Van Cappellen, P. & Y. Wang, 1996. Cycling of iron and manganese in surface sediments: A general theory for the coupled transport and reaction of carbon, oxygen, nitrogen, sulphur, iron and manganese. American Journal of Science 296: 197–243.
Winderlund, A., 1996. Early diagenetic remobilization of copper in near-shore marine sediments: a quantitative pore-water model. Marine Chemistry 54: 41–53.
Acknowledgements
The authors wish to thank their colleague, Miguel Quintans for helping with fieldwork.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Caetano, M., Madureira, MJ. & Vale, C. Exchange of Cu and Cd across the sediment-water interface in intertidal mud flats from Ria Formosa (Portugal). Hydrobiologia 587, 147–155 (2007). https://doi.org/10.1007/s10750-007-0673-y
Issue Date:
DOI: https://doi.org/10.1007/s10750-007-0673-y