
Abstract
Diabetic cardiomyopathy (DCM) is a diabetes mellitus–induced pathophysiological condition characterized by cardiac structural, functional, and metabolic changes that can result in heart failure (HF), in the absence of coronary artery disease, hypertension, and valvular heart disease. Metabolic alterations such as hyperglycemia, insulin resistance, hyperinsulinemia, and increased metabolism of free fatty acids result in oxidative stress, inflammation, advanced glycation end products formation, abnormalities in calcium homeostasis, and apoptosis that are responsible for structural remodeling. Cardiac stiffness, hypertrophy, and fibrosis eventually lead to dysfunction and HF with preserved ejection fraction and/or HF with reduced ejection fraction. In this review, we analyzed in detail the cellular and molecular mechanisms and the metabolic pathways involved in the pathophysiology of DCM. Different phenotypes are observed in DCM, and it is not clear yet if the restrictive and the dilated phenotypes are distinct or represent an evolution of the same disease. Phenotypic differences can be observed between T1DM and T2DM DCM, possibly explained by the different myocardial insulin action. Further studies are needed in order to better understand the underlying mechanisms of DCM and to identify appropriate therapeutic targets and novel strategies to prevent and reverse the progression toward heart failure in diabetic patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus and cardiovascular diseases
Diabetes mellitus (DM) has a current global prevalence of 463 million individuals, estimated to rise to 700 million people by 2045. DM represents the eight leading cause of death, and it is responsible for 11.3% of deaths globally [1].
DM significantly increases the risk of cardiovascular diseases (CVD). Patients with DM have a significantly higher risk of all-cause mortality and cardiovascular mortality. Patients with DM and poor glycemic control (hemoglobin A1c (HbA1c) > 9.7%/83 mmol/mol) have a cardiovascular mortality risk tenfold higher among patients with type 1 DM (T1DM) [2] and fivefold higher in subjects with type 2 DM (T2DM) [3]. CVD represent the main cause of morbidity and mortality in patients with DM, accounting for about two-thirds of overall deaths in patients with T2DM, with coronary artery disease (CAD) and ischemic cardiomyopathy as main contributors. According to the Framingham study, patients with DM have a twofold to fourfold increased risk of developing CAD and myocardial infarction (MI) and a fourfold to sixfold increased risk of developing congestive heart failure (HF). The incidence of HF results increased in both male and female diabetic patients, and this association is independent of CAD, hypertension, dyslipidemia, and obesity [4].
HF prevalence in patients with DM ranges from 19 to 26%. Each 1% increase in HbA1c is linked to a 30% increase in risk of HF in T1DM and 8% increase of risk of HF in T2DM [5].
Diabetic heart disease: definition
Diabetic heart disease includes CAD, cardiac autonomic neuropathy (CAN), and diabetic cardiomyopathy (DCM), often underdiagnosed [6] (Fig. 1).

Diabetic heart disease is a complex disease, including coronary artery disease (CAD), diabetic cardiomyopathy (DCM), and cardiac autonomic neuropathy (CAN)
CAD in patients with DM is characterized by rapid progression of disease, multivessel involvement, and lesions with more vulnerable features, often requiring coronary revascularization, with worse revascularization outcomes and 86% higher rates of in-stent restenosis [7].
CAN affects up to 65% patients with DM, it is a major cause of silent myocardial ischemia and MI, and it increases the risk of cardiac arrhythmias and sudden cardiac death. Initially, there is a subclinical stage with parasympathetic involvement, while toward the end stage of disease, there is sympathetic denervation and patients become symptomatic because of a poor response of heart rate and blood pressure to exercise and sleep, with consequent orthostatic hypotension, reduced exercise tolerance, and non-dipping or reverse dipping at night [8].
DCM is a DM-induced pathophysiological condition characterized by cardiac structural, functional, and metabolic changes that can result in HF, in the absence of CAD, hypertension, and valvular heart disease. DCM is generally asymptomatic in the initial stages. DCM was traditionally described with an earlier stage characterized by left ventricle (LV) hypertrophy, stiffness, and decreased LV compliance characterized by reduced early diastolic filling, increased atrial filling, and prolonged isovolumetric relaxation time, and a later stage characterized by cardiac fibrosis, LV dilation, and systolic dysfunction, with the onset of HF symptoms [9]. More recently, it was described a restrictive pattern with preserved ejection fraction (EF) and a dilated pattern with reduced EF [10]. Therefore, DCM may lead to both HF with preserved EF (HFpEF) and with reduced EF (HFrEF).
Studies that compared equal size myocardial infarct areas in non-diabetic and diabetic patients found out that the incidence of HF is statistically significant higher in the diabetic patients group than in those without DM, suggesting that ischemic and diabetic cardiomyopathy are interrelated entities, amplifying maladaptive contractile effects in DM patients [11].
Potential damage of hyperglycemia on cardiac muscle and coronary vessels
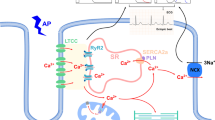
Metabolic alterations, such as hyperglycemia, insulin resistance, and increased metabolism of free fatty acids (FFA), result in oxidative stress, inflammation, advanced glycation end products (AGEs) formation, abnormalities in calcium homeostasis, apoptosis, and fibrosis. These changes are responsible for cardiac remodeling and dysfunction [12] (Fig. 2). One of the most important mechanisms involved in the development of DCM is chronic hyperglycemia, which induces nonenzymatic glycation of proteins, lipids, and lipoproteins resulting in formation of AGEs, altering their functional properties [13].

Metabolic alterations in diabetic cardiomyopathy
The formation of AGEs on vascular cells and myocytes causes cross-linking of collagen molecules to each other. This leads to the loss of collagen elasticity and degradation, to an increase in cardiac interstitial and perivascular fibrosis, and subsequently to a reduction of arterial and myocardial compliance [14]. This can also lead to coronary microvascular stenosis and microaneurysms.
Moreover, AGEs interact and upregulate their receptors RAGE (Receptors for AGE). This interaction activates transcription factors, such as nuclear factor-κB (NFκB) and its target genes, with production of pro-inflammatory cytokines, such as interleukins (IL-1β, IL-6, IL-18), tumor necrosis factor (TNF-α), and myeloperoxidases, responsible for cardiac damage [15]. NF-kB activation is promoted not only by AGE-RAGE’s interactions, but also directly by reactive oxygen species (ROS) and by the renin–angiotensin–aldosterone system (RAAS) activation. Hyperglycemia also plays a key role in the development of myocardial fibrosis, because of increased transcription of collagen by enhanced expression of vascular growth factors (VGF) and tissue growth factor beta 1 (TGF-β1), with dysregulation of extracellular matrix degradation. Cardiac fibrosis leads to increased LV stiffness and decreased ventricular wall compliance, resulting in both systolic and in particular diastolic dysfunction [16]. Moreover, high glucose concentrations increase ROS. Increased levels of superoxide anion react with nitric oxide (NO) released by endothelial nitric oxide synthase (eNOS). The lower bioavailability of myocardial NO reduces cyclic guanosine monophosphate (cGMP) with decreased protein kinase G (PKG) activity. PKG activity reduction in cardiomyocytes causes titin hypophosphorylation, which controls diastolic myocardial distensibility [17]. In addition, in a healthy heart, the contraction is mediated by calcium (Ca2+) entrance through L-type Ca2+ channels. This triggers the release of Ca2+ from the sarcoplasmic reticulum via ryanodine receptors (RyR), leading to interaction between actin and myosin filaments. The relaxation occurs when Ca2+ is removed from the cytosol, and it is brought into the sarcoplasmic reticulum by the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) 2a [18]. In DCM, RyR activity is damaged by oxidative stress, and there is a decrease in SERCA2a protein levels; therefore, the final result is an increase in intracellular calcium (Ca2+) and a decrease in sarcoplasmic Ca2+ uptake [11]. These alterations are responsible for cardiac diastolic stiffness and dysfunction in DCM [19].
Finally, hyperglycemia and lipotoxicity raise protein kinase C (PKC) activity in fibroblast, with deposition of collagen and fibrosis [20]. PKC signaling pathways are also induced by oxidative stress, inflammation and RAAS, and sympathetic nervous system (SNS) activity. The isoforms alpha, beta, delta, epsilon, and theta of PKC have been proposed to contribute to the development of diabetic cardiac hypertrophy [21].
Metabolic pathways in the pathophysiology of DCM
Under normal conditions, cardiomyocytes generate energy mainly by fatty acid oxidation, with a small contribution from glucose that increases under stressful situations. Fatty acid enters cardiomyocytes through CD36 (also referred to as scavenger receptor B2), while glucose through GLUT-4 [22].
Insulin resistance is associated with increased CD36-mediated fatty acid uptake and decreased AMP-activated protein kinase (AMPK) activation and consequent reduced GLUT-4 expression and translocation to the plasma membrane, reduced GLUT4-mediated glucose uptake, and reduced fatty acids oxidation and glycolysis. Chronic hyperglycemia and hyperinsulinemia are associated with increased ROS generation and oxidative stress that diverts glucose metabolism from glycolytic pathway to alternative pathways such as hexosamine pathway with enzymatic O-GlcNAcylation of cardiomyocyte proteins and polyol pathway, with AGEs generation.
AGEs accumulation lead to AGE/RAGE interaction, extracellular matrix (ECM) remodeling with collagen-elastin cross-linkage, NF-kB signaling, pro-inflammatory cytokines and TGF-beta production, increased ROS production and cardiac oxidative stress, depressed sarcoplasmic reticulum (SR) function with reduced Ca2+ reuptake into SR, shift in myosin heavy chain (MHC) from alpha-MHC to beta-MHC, atrial natriuretic peptide (ANP), and brain natriuretic peptide (BNP) upregulation. These alterations lead to myocyte hypertrophy and myocardial fibrosis/stiffness.
The increased fatty acid uptake via CD-36 leads to increased fatty acid oxidation that exceeds mitochondrial oxidative capacity and determines mitochondria dysfunction, with reduced ATP production and altered myocardial Ca2+ handling. High glucose exposure determines decreased transcriptional expression of mito calcium uniporter (MCU) and of MCU-bracketing protein EMRE [23]. These changes determine reduced myocardial contractility, endothelial damage with microvascular dysfunction, increased ROS and oxidative stress, NO destruction and reduced bioavailable NO, pro-inflammatory cytokines and lipotoxic metabolites production, and induced lipoapoptosis of cardiomyocyte and myocardial fibrosis. High levels of fatty acids, hyperglycemia, and impaired insulin metabolic signaling activate the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome assembly and procaspase-1 activation that processes IL-1beta and IL-18 precursors and enhances NF-kB pathway [24].
The hexosamine pathway upregulation with sustained increased mitochondrial O-GlcNAc levels determines post-translational modification of cardiac proteins and consequent altered myocardial calcium handling with contractile dysfunction and heart failure.
Insulin resistance, hyperinsulinemia, and hyperglycemia also cause activation of RAAS, increased angiotensin II activity, vascular resistance, and aldosterone activity that lead to cardiomyocyte hypertrophy, hypertension, and increased cardiac fibroblast proliferation.
Insulin resistance also decreases insulin-stimulated coronary e-NOS activity and NO production, reducing NO coronary vasodilation and insulin-mediated capillary recruitment that lead to an impaired delivery of insulin and glucose necessary for normal myocardial energetics. The result is a reduction of sarcoplasmic reticulum Ca + uptake. Impairment of NO production also leads to phosphorylation of titin increasing the ratio of still titin isoform expression and to increased activation of collagen cross-linking enzymes and fibrosis.
Hyperinsulinemia induce cardiomyocyte hypertrophy by binding to insulin-like growth factor 1 (IGF-1) receptor. IGF1 produced by cardiomyocytes can also stimulate cardiomyocyte hypertrophy through the insulin receptor, extracellular signal-regulated kinase 1/2, and phosphatidylinositol 3-kinase pathways [25].
SGLT-2 expression is significantly increased in diabetic patients, with consequent glomerular hyperfiltration, increased reabsorption of glucose, and elevated plasma glucose levels [26].
In the pathogenesis of DCM have also been implicated PKC signaling pathways, that promote cardiac hypertrophy and fibrosis, and MAPK and JNK activation pathway, that contribute to oxidative stress, endoplasmic reticulum stress and interstitial fibrosis [27], and increased cardiomyocyte apoptosis.
Several lipid metabolites contribute to exacerbating DCM by impairing insulin metabolic signaling. Diacylglycerols accumulation in the plasma membrane can activate PKC ε inducing insulin resistance and reducing NO production, while ceramides can directly activate PKCs and attenuate GLUT4 translocation and glucose uptake [5].
At nuclear level, DCM pathophysiology includes repression of NF-E2-related factor 2 (Nrf2) and activation of NF-kB and CAMP-responsive element modulator (CREM). The repression of Nrf2 expression promotes the expression of antioxidant proteins in response to oxidative stress, such as hemoxygenase. NF-kB is a transcriptional factor that promotes the expression of pro-inflammatory cytokines and profibrotic genes. CREM is a transcriptional factor that promotes cardiac fibrosis [28] and may also promote epigenetic modifications in cardiac proteins.
Finally, also exosomes and miRNA abnormalities may play a role in DCM. Exosomes released from cardiomyocytes of DCM patients contain high levels of miR320 and are transported to coronary endothelial cells, leading to a decreased NO production. Different miRNAs have been reported to be increased in T2DM and T1DM and were found to be involved in insulin sensitivity, cardiomyocyte hypertrophy, fibrosis, and diastolic dysfunction [5]. Decreased levels of miR-494-3p upregulate activator protein 1 (AP-1) JunD activity that promotes PPARgamma-dependent genes (such as CD-36, FAS, LpL) transcription, involved in myocardial lipid uptake, hydrolysis, and storage, therefore leading to cardiac steatosis, lipotoxic damage, and metabolic cardiomyopathy [29]. miR-122-5p, which targets the metalloproteinases MMP-16 and MMP-2, their regulator (tissue inhibitor of MMPs), and the ECM through MMP-2 modulation, results upregulated in T2DM. Increased levels of miR-122-5p downregulate MMP-2 activity that is involved in ECM rearrangement. Subendocardial fibrosis leads to increased ventricular torsion, due to defective shortening of the fragile subendocardial fibers. The torsion eventually reaches a plateau and exhaust its compensatory role and decouples from strain that appears reduced in all stages of DCM. Within 5 years of DCM onset, cardiac magnetic resonance (CMR) assessment demonstrated that the progression of increasing cardiac hypertrophy is associated with progressive impairment in strain, depletion of the compensatory role of torsion, and changes in viscoelastic contraction dynamics. Cardiac remodeling reduces the potential energy stored during the systole, leading to a shorter and less effective systolic phase. Also the diastolic phase is less effective, with a reduced recoil rate and an impaired isovolumic relaxation. The progression toward LV dilation accompanied by an increased cardiac hypertrophy is independent from glycemic control [30]. Except in the very early stage, strict glycemic control was found to be not sufficient to revert the pathological cardiac processes in DCM [31].
DCM finally leads LV remodeling, cardiac diastolic and systolic dysfunction, and endothelial damage with microvascular dysfunction [6].
Cell death in DCM
Molecular mechanisms that increase fibrosis and myocardial inflammation in diabetic patients can activate both pro-apoptotic and necrotic cell death signaling pathways [32].
Diabetes increases apoptosis of myocytes by an 85-fold and necrosis in myocytes by fourfold. Also endothelial cells and fibroblasts apoptosis and necrosis rate are significantly increased in DM patients [33].
Potential mechanisms underlying the increased cell death in DM patients are leptin deficiency, hyperglycemia through a Rac 1-mediated increase in nicotinamide adenine dinucleotide phosphate (NADPH) and mitochondrial ROS production and activation of RAAS [34].
In DCM we observe a decreased AMPK activity and consequent increased mammalian target of rapamycin (m-TOR) signaling pathway that leads to a reduction in cardiomyocyte autophagy and consequent exacerbated cardiac dysfunction and apoptosis. Autophagy is a cytoprotective mechanism; indeed a reduced cardiac autophagic activity results in the accumulation of clustered and damaged mitochondria and polyubiquitinated proteins that induce respectively the release of ROS and pro-apoptotic factors such as cytochrome c and an increase in endoplasmic reticulum stress, which both lead to cardiomyocyte apoptosis [35].
Phenotype adaptation according to myocyte and vascular cell alterations in DCM
In 1954, Lundbaek was the first to describe a specific vascular disease of long-standing diabetes, named diabetic angiopathy [36]. Almost 20 years later, in 1972, Rubler et al. identified a new type of cardiomyopathy in diabetic patients characterized by myocardial hypertrophy, fibrosis, and diabetic microangiopathy, in the absence of major CAD [37]. Two years later, the Framingham study established the increasing risk of HF in the diabetic patients [38]. In 2011, Maisch et al. divided DCM into four stages: stage 1 included diastolic dysfunction with normal ejection fraction, often associated with hypertrophy; stage 2 included diastolic and systolic dysfunction with reduced ejection fraction. Both stages were not affected by CAD, valvular heart disease, and hypertension. Stage 3 included DCM with diastolic and systolic dysfunction with involvement of microvascular disease and/or microbial infection and/or inflammation and/or hypertension but without CAD, and stage 4 included DCM with HF that may also be attributed to clinical infarction or ischemia [39].
Nowadays, the improvement in non-invasive instrumental examinations, such as echocardiography and cardiac magnetic resonance, made it possible to learn more about the structural alterations of hearts affected by DM. DM causes concentric remodeling, both concentric and eccentric hypertrophy, increases left ventricular mass and wall thicknesses and finally causes contractile dysfunction of the LV [40, 41].
DCM was initially described by Rubler et al. as dilated phenotype with eccentric left ventricular remodeling and systolic dysfunction known as HFrEF [37]. However, in the last few years, a new distinct phenotype from dilated to concentric hypertrophic pattern has been recognized: the so called emerging restrictive phenotype. It is characterized by normal LV diameters and increased wall thickness, elevated LV filling pressures, diastolic dysfunction, and HFpEF. Current paradigm arises few unanswered questions: (1) whether the two phenotypes (restrictive/HFpEF and dilated/HFrEF) are distinct or rather an evolution of the same disease; (2) if each subtype subtends different energetic cell dysfunction and metabolic derangement; and (3) the cardiovascular risk associated with distinct phenotype and the related therapeutic strategies. However, a universal DCM definition based on translational data and epidemiologic and prognostic features is still lacking, and several doubts remain about the real expression of cardiac remodeling, the influence of associated macro or microvascular CAD, and the possible mechanisms for the transition from one to the other pattern.
Seferovic and Paulus were the first authors purposing this distinction arguing that DCM evolves as two independent morphological phenotypes: restrictive, linked to coronary microvascular endothelial dysfunction and prevalent in T2DM, and dilated, associated with cardiomyocyte cell death and more common in T1DM patients [10].
The main features observed in the two phenotypes are due to interactions and pathophysiological alterations between endothelial cells, cardiomyocytes, and fibroblasts (Fig. 3). In the restrictive phenotype, cardiomyocyte hypertrophy and reactive fibrosis are the consequence of these alterations, with HFpEF development. First of all, hyperglycemia, lipotoxicity, and AGEs increase mitochondrial ROS in endothelial cells, which determines lower bioavailability of endothelial NO and consequently decreases PKG activity in adjacent cardiomyocytes leading to hypertrophy, stiffness, and LV diastolic dysfunction. In fibroblasts hyperglycemia and lipotoxicity raise PKC activity resulting in collagen deposition and reactive interstitial fibrosis [12]. Instead, the dilated phenotype is characterized by cardiomyocytes cell death and fibrosis replacement, leading to HFrEF pattern. One of the most important mechanisms is ischemic injury because of microvascular rarefaction, AGEs, and autoimmunity-related inflammatory cells. Tissue hypoxia leads to increased ROS and cardiomyocyte cell death resulting from oxidative stress. Hyperglycemia and lipotoxicity are responsible for replacement fibrosis deposition, through increased PKC activity in fibroblast. Lastly, a key contributing role in development and progression of myocardial injury and LV dysfunction is played by inflammatory cytokines such as IL-1β, IL-6, TNF-α, and TGF-β1 and the inflammatory transcriptional regulator NFκB, leading to dilated CMD [42, 43]. Therefore, the restrictive/HFpEF phenotype is characterized by hypertrophied cardiomyocytes with preserved sarcomeric structure and collagen deposition in-between cardiomyocytes, while in the dilated/HFrEF phenotype, cardiomyocytes are small and damaged, sarcomeric structure disappeared, and collagen deposition covers larger areas.

Morphological phenotypes in diabetic cardiomyopathy
Epigenetic modifications have an important role in regulating the pathways involved in HF. DNA methylation, histone modifications, and non-coding RNAs play a key role in cardiac fibrosis, hypertrophic remodeling, myocardial stiffness, and vascular remodeling, through reprogramming of gene expression and reactivation of fetal cardiac genes. Specific epigenetic patterns allow to identify patients with HF and also to discriminate between different HF phenotypes. Hypomethylations of CTGF and MMP-2 are potential epigenetic biomarkers in HFpEF, due to their strong involvement in cardiac fibrosis. Histone modifications H3K4me3, H3K9me3, and H3K36me3 are causal biomarkers of LV hypertrophy and remodeling in HFpEF and can be used for a direct, personalized intervention. miR-183-3p is downregulated in both HFrEF and HFpEF. miR-190a could be useful to detect HFpEF and to discriminate between HFpEF and HFrEF but requires further validation [44].
The differentiation between restrictive/HFpEF and dilated/HFrEF phenotypes has important clinical consequences and therapeutic implications related to CV and metabolic management. When dilated pattern occurs, according to the heart failure guidelines, the traditional algorithm of HFrEF treatment can be applied: angiotensin-converting enzyme inhibitor (ACE-I), beta-blocker, angiotensin receptor blocker (ARB), aldosterone antagonists, ivabradine, angiotensin receptor neprilysin inhibitor (ARNI), and resynchronization therapy [45].
Unfortunately, most of diabetic patients develop a hypertrophic adaptation with restrictive pattern, probably because diabetes is often associated with hypertension and other metabolic disorders. In the latter case, therapeutic options are restricted to the monitorization of CV risk factors and glycemic control, without any evidence of benefit on both risk of HF and CV mortality reduction. Current weaknesses involve also the hypoglycemic treatment that is mostly associated with mild risk reduction and scarce impact in HF development particularly with traditional agents. The concern of HF became even more relevant considering that 2/3 of these patients develop HFpEF in which the role of both cardiovascular and antidiabetic drugs is neutral or unexplored. However, despite the different response to medical therapy, HFrEF and HFpEF have a similar prognosis [46].
Sodium-glucose co-transporter-2 (SGLT2) inhibitors had deeply changed the natural history of diabetic HF, reducing CV mortality and HF hospitalizations. Left ventricular hypertrophy (LVH) is a negative prognostic marker in patients with HFpEF. DAPA-LVH trial and EMPA-HEART Cardiolink-6 trial showed the beneficial effect of SGLT2 inhibitor dapagliflozin and empaglifozin, respectively, on LVH regression (assessed as LV mass regression by cardiac MRI) in patients with T2DM. The underlying mechanisms proposed for SGLT2 inhibitor-mediated LVH regression are decreased blood pressure and afterload, reduced visceral adipose tissue, improved insulin sensitivity, and reduced levels of systemic inflammation, prevention of cardiomyocytes and endothelial cells dysfunction, and improvement of diastolic dysfunction [47].
Several drugs targeting epigenetic modifications underlying HFpEF have been developed (“epidrugs”), and some have already been approved by the Food and Drug Administration (FDA). Folates restore promoter CpG methylation of different genes regulating endothelial function, NO bioavailability, adipogenesis, and oxidative stress pathways implicated in HFpEF. Natural compounds such as sulforaphane (contained in broccoli sprouts) and epigallocatechin-3-gallate (found in green tea) improve microvascular endothelial function. Histone deacetylase inhibitor Vorinostat prevents pathological cardiac hypertrophic remodeling and diastolic dysfunction. MicroRNA therapeutics are currently under development in preclinical and clinical trials [44].
DCM in T1DM versus T2DM
The phenotypes and underlying mechanisms of DCM have been mostly investigated in T2DM animals and humans, while the impact of T1DM on diastolic and systolic impairment is less clear [5], because results of human studies remain controversial and the metabolic derangements and the phenotype may be attenuated or masked by the fact that patients are treated with insulin. The underlying mechanisms and clinical features of DCM in T1DM and T2DM probably overlap, but some differences were observed in phenotypes [48], such as cardiomyocyte autophagy, increased in T1DM and suppressed in T2DM [49], systolic function, generally preserved in T1DM Akita diabetic mice, and cardiac hypertrophy, not observed in T1DM Akita diabetic mice [50]. These phenotypic differences may be explained by differences in myocardial insulin action, since T1DM is characterized by insulin deficiency while T2DM by insulin resistance with hyperinsulinemia, and this could have effects on cell survival, cell growth, and other cellular pathways. Glycemic control reduces the prevalence of DCM and of CVD. In T1DM rodent models normalization of glycemic values through insulin replacement was associated with reduced myocardial hypertrophy, collagen content, and diastolic dysfunction [51]. In rats, chronic diabetes is associated with a shift in cardiac myosin heavy chain from V1 to V3 isoforms that correlates with depressed contractility, reversible with insulin treatment [52].
Conclusions
DM significantly increases the risk of heart disease. Diabetic heart disease is a complex disease, represented by three clinical entities: CAD, CAN, and DCM. This review focused on the metabolic, structural, and functional changes in the myocardium that occur in DCM, that is, a pathophysiological condition characterized by cardiac structural, functional, and metabolic changes that can result in HF, in the absence of CAD, hypertension, and valvular heart disease.
Hyperglycemia, systemic insulin resistance, and hyperinsulinemia are the key etiological factors in the development of DCM, inducing impaired cardiac insulin signaling, increased levels of FFA and growth factors, impaired substrate utilization and lipid metabolism, and altered calcium homeostasis. Structural changes are mainly represented by cardiac stiffness, hypertrophy, and fibrosis that eventually lead to HFpEF and/or HFrEF.
The signaling pathways underlying DCM pathophysiological events include decreased AMPK activity, increased PKC activity, sustained increase of O-GlcNAcylation, increased MAPK and SGLT2 function, and dysregulation of exosomes and of miRNA. At nuclear level, it was demonstrated an increase of CREM expression and NF-kB signaling and a reduction of Nrf-2 expression. The pathophysiological process of DCM involves mitochondria dysfunction, impairment of mitochondria Ca2+ handling, inflammation, ROS production and oxidative stress, AGEs-RAGE interactions, reduced bioavailability of NO, activation of RAAS, ER stress, autonomic neuropathy, lipotoxicity, cardiomyocyte death, and microvascular dysfunction. Some mechanisms, such as autophagy and miRNA require further study.
Traditionally it has been described an initial stage of DCM, clinically asymptomatic and characterized by cardiac stiffness, hypertrophy, and fibrosis, resulting in diastolic dysfunction, eventually evolving in LV dilation, systolic dysfunction, and symptomatic heart failure. More recently, a new distinct phenotype has been recognized besides the dilated phenotype with HFrEF: the restrictive phenotype, characterized by normal LV dimensions, diastolic dysfunction, and HFpEF.
Glycemic control reduces the prevalence of DCM and of CVD, but glycemic control alone is not sufficient to prevent diabetic heart disease development.
A universal definition of DCM based on translational data and epidemiologic and prognostic features is still lacking. Further investigations are needed in order to understand potential differences in underlying mechanisms and phenotypes for DMC in T1DM and T2DM patients. It is not clear yet whether the two described phenotypes (restrictive/HFpEF and dilated/HFrEF) are distinct or rather an evolution of the same disease and what are the possible underlying metabolic mechanisms for the transition from one phenotype to the other. Greater efforts should be made to understand the precise molecular mechanisms involved in the initiation and progression of DCM and in order to identify potential appropriate therapeutic targets and novel pharmacological strategies that may help to prevent and reverse the progression toward HF in diabetic patients. The result of large epigenomic studies in the upcoming future will help to define the links between genetics, epigenetic, and HF and validate epigenetic targeted personalized therapies.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
International Diabetes Federation (2019) IDF Diabetes Atlas, 9th edition (Online). [Cited November 2, 2020]. Available from: https://www.diabetesatlas.org/en
Lind M, Svensson AM, Rosengren A (2015) Glycemic control and excess mortality in type 1 diabetes. N Engl J Med 372(9):880–881. https://doi.org/10.1056/NEJMc1415677 (PMID: 25714168)
Tancredi M, Rosengren A, Svensson AM, Kosiborod M, Pivodic A, Gudbjörnsdottir S, Wedel H, Clements M, Dahlqvist S, Lind M (2015) Excess mortality among persons with type 2 diabetes. N Engl J Med 373:1720–1732 [PMID: 26510021 https://doi.org/10.1056/NEJMoa1504347]
Kannel WB, McGee DL (1979) Diabetes and cardiovascular disease. The Framingham study. JAMA 241:2035–2038 [PMID: 430798 https://doi.org/10.1001/jama.241.19.2035]
Jia G, Hill MA, Sowers JR (2018) Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 122:624–638 (PubMed: 29449364)
Rajbhandari J, Fernandez CJ, Agarwal M, Yeap BXY, Pappachan JM (2021) Diabetic heart disease: a clinical update. World J Diabetes 12(4):383–406
Cassese S, Byrne RA, Schulz S, Hoppman P, Kreutzer J, Feuchtenberger A, Ibrahim T, Ott I, Fusaro M, Schunkert H, Laugwitz KL, Kastrati A (2015) Prognostic role of restenosis in 10 004 patients undergoing routine control angiography after coronary stenting. Eur Heart J 36:94–99 [PMID: 25298237 https://doi.org/10.1093/eurheartj/ehu383]
Spallone V (2019) Update on the impact, diagnosis and management of cardiovascular autonomic neuropathy in diabetes: what is defined, what is new, and what is unmet. Diabetes Metab J 43:3–30 [PMID: 30793549 https://doi.org/10.4093/dmj.2018.0259]
Jia G, DeMarco VG, Sowers JR (2016) Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 12:144–153. https://doi.org/10.1030/nrendo.2015.216
Seferović PM, Paulus WJ (2015) Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J 36(27):1718–27, 1727a-1727c. https://doi.org/10.1093/eurheartj/ehv134. Epub 2015 Apr 17. PMID: 25888006
Dillmann WH (2019) Diabetic Cardiomyopathy. Circ Res 124(8):1160–1162. https://doi.org/10.1161/CIRCRESAHA.118.314665.PMID:30973809;PMCID:PMC6578576
Evangelista I, Nuti R, Picchioni T, Dotta F, Palazzuoli A (2019) Molecular dysfunction and phenotypic derangement in diabetic cardiomyopathy. Int J Mol Sci 20(13):3264. https://doi.org/10.3390/ijms20133264.PMID:31269778;PMCID:PMC6651260
Goldin A, Beckman JA, Schmidt AM, Creager MA (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114:597–605
Aronson D (2003) Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J Hypertens 21(1):3–12
Aragno M, Mastrocola R, Medana C, Catalano MG, Vercellinatto I, Danni O, Boccuzzi G (2006) Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology 147(12):5967–5974. https://doi.org/10.1210/en.2006-0728 (Epub 2006 Aug 24 PMID: 16935841)
Li CJ, Lv L, Li H, Yu DM (2012) Cardiac fibrosis and dysfunction in experimental diabetic cardiomyopathy are ameliorated by alpha-lipoic acid. Cardiovasc Diabetol 19(11):73. https://doi.org/10.1186/1475-2840-11-73.PMID:22713251;PMCID:PMC3472273
Kaludercic N, Di Lisa F (2020) Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med 18(7):12. https://doi.org/10.3389/fcvm.2020.00012.PMID:32133373;PMCID:PMC7040199
Bertero E, Maack C (2018) Calcium signaling and reactive oxygen species in mitochondria. Circ Res 122(10):1460–1478. https://doi.org/10.1161/CIRCRESAHA.118.310082 (PMID: 29748369)
Talukder MA, Kalyanasundaram A, Zuo L, Velayutham M, Nishijima Y, Periasamy M, Zweier JL (2008) Is reduced SERCA2a expression detrimental or beneficial to postischemic cardiac function and injury? Evidence from heterozygous SERCA2a knockout mice. Am J Physiol Heart Circ Physiol 294:H1426–H1434. https://doi.org/10.1152/ajpheart.01016.2007
Way KJ, Isshiki K, Suzuma K, Yokota T, Zvagelsky D, Schoen FJ, Sandusky GE, Pechous PA, Vlahos CJ, Wakasaki H, King GL (2002) Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C beta2 activation and diabetes. Diabetes 51:2709–2718
Lei S, Li H, Xu J et al (2013) Hyperglycemia-induced protein kinase C β2 activation induces diastolic cardiac dysfunction in diabetic rats by impairing caveolin-3 expression and Akt/eNOS signaling. Diabetes 62(7):2318–2328. https://doi.org/10.2337/db12-1391
Palomer X, Pizarro-Delgado J, Vázquez-Carrera M (2018) Emerging actors in diabetic cardiomyopathy: heartbreaker biomarkers or therapeutic targets? Trends Pharmacol Sci 39:452–467 [PMID: 29605388 https://doi.org/10.1016/j.tips.2018.02.010]
Granatiero V, De Stefani D, Rizzuto R (2017) Mithocondrial Calcium Handling in Physiology and Disease. Adv Exp Med Biol 982:25–47. https://doi.org/10.1007/978-3-319-55330-6_2
Pal PB, Sonowal H, Shukla K, Srivastava SK, Ramana KV (2017) Aldose reductase mediates NLRP3 inflammasome-initiated innate immune response in hyperglycemia-induced Thp1 monocytes and male mice. Endocrinology 158(10):3661–3675. https://doi.org/10.1210/en.2017-00294.PMID:28938395;PMCID:PMC5659696
Sundgren NC, Giraud GD, Schultz JM, Lasarev MR, Stork PJ, Thornburg KL (2003) Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am J Physiol Regul Integr Comp Physiol 285(6):R1481–R1489. https://doi.org/10.1152/ajpregu.00232.2003 (Epub 2003 Aug 28 PMID: 12947030)
von Lewinski D, Rainer PP, Gasser R, Huber MS, Khafaga M, Wilhelm B, Haas T, Mächler H, Rössl U, Pieske B (2010) Glucose-transporter-mediated positive inotropic effects in human myocardium of diabetic and nondiabetic patients. Metabolism 59(7):1020–1028. https://doi.org/10.1016/j.metabol.2009.10.025 (Epub 2009 Dec 31 PMID: 20045149)
Wang Y, Zhou S, Sun W, McClung K, Pan Y, Liang G, Tan Y, Zhao Y, Liu Q, Sun J, Cai L (2014) Inhibition of JNK by novel curcumin analog C66 prevents diabetic cardiomyopathy with a preservation of cardiac metallothionein expression. Am J Physiol Endocrinol Metab 306(11):E1239–E1247. https://doi.org/10.1152/ajpendo.00629.2013 (Epub 2014 Apr 8 PMID: 24714399)
Barbati SA, Colussi C, Bacci L, Aiello A, Re A, Stigliano E, Isidori AM, Grassi C, Pontecorvi A, Farsetti A, Gaetano C, Nanni S (2017) Transcription factor CREM mediates high glucose response in cardiomyocytes and in a male mouse model of prolonged hyperglycemia. Endocrinology 158(7):2391–2405. https://doi.org/10.1210/en.2016-1960 (PMID: 28368536)
Costantino S, Akhmedov A, Melina G, Mohammed SA, Othman A, Ambrosini S, Wijnen WJ, Sada L, Ciavarella GM, Liberale L, Tanner FC, Matter CM, Hornemann T, Volpe M, Mechta-Grigoriou F, Camici GG, Sinatra R, Lüscher TF, Paneni F (2019) Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur Heart J 40(12):997–1008. https://doi.org/10.1093/eurheartj/ehy903 (PMID: 30629164)
Pofi R, Giannetta E, Galea N, Francone M, Campolo F, Barbagallo F, Gianfrilli D, Venneri MA, Filardi T, Cristini C, Antonini G, Badagliacca R, Frati G, Lenzi A, Carbone I, Isidori AM (2021) Diabetic cardiomiopathy progression is triggered by miR122-5p and involves extracellular matrix: a 5-year prospective study. JACC Cardiovasc Imaging 14(6):1130–1142. https://doi.org/10.1016/j.jcmg.2020.10.009 (Epub 2020 Nov 18 PMID: 33221242)
Boussageon R, Bejan-Angoulvant T, SaadatianElahi M et al (2011) Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. BMJ 343:d4169
Bugger H, Abel ED (2014) Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57(4):660–671. https://doi.org/10.1007/s00125-014-3171-6
Frustaci A, Kajstura J, Chimenti C et al (2000) Myocardial cell death in human diabetes. Circ Res 87:1123–1132
Boudina S, Abel ED (2010) Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 11(1):31–39. https://doi.org/10.1007/s11154-010-9131-7
Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH (2011) Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60(6):1770–8. https://doi.org/10.2337/db10-0351. Epub 2011 May 11. PMID: 21562078; PMCID: PMC3114402
Lundbaek K (1954) Diabetic angiopathy: a specific vascular disease. Lancet 266(6808):377–379. https://doi.org/10.1016/s0140-6736(54)90924-1 (PMID: 13131862)
Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A (1972) New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 30(6):595–602. https://doi.org/10.1016/0002-9149(72)90595-4 (PMID: 4263660)
Kannel WB, Hjortland M, Castelli WP (1974) Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 34(1):29–34. https://doi.org/10.1016/0002-9149(74)90089-7 (PMID: 4835750)
Maisch B, Alter P, Pankuweit S (2011) Diabetic cardiomyopathy–fact or fiction? Herz 36(2):102–115. https://doi.org/10.1007/s00059-011-3429-4 (PMID: 21424347)
Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER, Howard BV (2000) Impact of diabetes on cardiac structure and function: the strong heart study. Circulation 101(19):2271–2276. https://doi.org/10.1161/01.cir.101.19.2271 (PMID: 10811594)
Levelt E, Mahmod M, Piechnik SK, Ariga R, Francis JM, Rodgers CT, Clarke WT, Sabharwal N, Schneider JE, Karamitsos TD, Clarke K, Rider OJ, Neubauer S (2016) Relationship between left ventricular structural and metabolic remodeling in type 2 diabetes. Diabetes 65(1):44–52. https://doi.org/10.2337/db15-0627. Epub 2015 Oct 5. PMID: 26438611; PMCID: PMC4890658
MinW, Bin ZW, Quan ZB, Hui ZJ, Sheng FG (2009) The signal transduction pathway of PKC/NF-kappa B/c-fos may be involved in the influence of high glucose on the cardiomyocytes of neonatal rats. Cardiovasc Diabetol 8:8
Levine B, Kalman J, Mayer L, Fillit HM, Packer M (1990) Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 323:236–241
Hamdani N, Costantino S, Mügge A, Lebeche D, Tschöpe C, Thum T, Paneni F (2021) Leveraging clinical epigenetics in heart failure with preserved ejection fraction: a call for individualized therapies. Eur Heart J 42(20):1940–1958. https://doi.org/10.1093/eurheartj/ehab197 (PMID: 33948637)
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P (2016) Wytyczne ESC dotyczące diagnostyki i leczenia ostrej i przewlekłej niewydolności serca w 2016 roku [2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure]. Kardiol Pol 74(10):1037–1147. Polish. https://doi.org/10.5603/KP.2016.0141. PMID: 27748494
Park JJ (2021) Epidemiology, pathophysiology, diagnosis and treatment of heart failure in diabetes. Diabetes Metab J 45(2):146–157. https://doi.org/10.4093/dmj.2020.0282. Epub 2021 Mar 25. PMID: 33813813; PMCID: PMC8024162
Paneni F, Costantino S, Hamdani N (2020) Regression of left ventricular hypertrophy with SGLT2 inhibitors. Eur Heart J 41(36):3433–3436. https://doi.org/10.1093/eurheartj/ehaa530 (PMID: 32620974)
Hölscher ME, Bode C, Bugger H (2016) Diabetic cardiomyopathy: does the type of diabetes matter?. Int J Mol Sci 17(12):2136. Published 2016 Dec 18. https://doi.org/10.3390/ijms17122136
Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, Watanabe T, Morishita K, Okada H, Kawasaki M, Seishima M, Minatoguchi S (2015) Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 11(7):1146–1160. https://doi.org/10.1080/15548627.2015.1051295.PMID:26042865;PMCID:PMC4590644
Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, Yun UJ, McQueen AP, Wayment B, Litwin SE, Abel ED (2008) Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes 57(11):2924–32. https://doi.org/10.2337/db08-0079. Epub 2008 Aug 4. PMID: 18678617; PMCID: PMC2570388
Tate M, Deo M, Cao AH, Hood SG, Huynh K, Kiriazis H, Du XJ, Julius TL, Figtree GA, Dusting GJ, Kaye DM, Ritchie RH (2017) Insulin replacement limits progression of diabetic cardiomyopathy in the low-dose streptozotocin-induced diabetic rat. Diab Vasc Dis Res 14(5):423–433. https://doi.org/10.1177/1479164117710390 (Epub 2017 May 31 PMID: 28565941)
Pollack PS, Malhotra A, Fein FS, Scheuer J (1986) Effects of diabetes on cardiac contractile proteins in rabbits and reversal with insulin. Am J Physiol 251(2 Pt 2):H448–H454. https://doi.org/10.1152/ajpheart.1986.251.2.H448 (PMID: 2943166)
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Prandi, F.R., Evangelista, I., Sergi, D. et al. Mechanisms of cardiac dysfunction in diabetic cardiomyopathy: molecular abnormalities and phenotypical variants. Heart Fail Rev 28, 597–606 (2023). https://doi.org/10.1007/s10741-021-10200-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-021-10200-y