
Abstract
Acute heart failure hospitalizations complicated by diuretic resistance are associated with worse outcomes. Yet, quantification of the frequency and accompanying risk from loop diuretic resistance is limited by the absence of a comprehensive definition with universal clinical application. Herein, we outline limitations of the current metrics used to identify and define diuretic resistance. We discuss the best available methods to identify and prognosticate outcomes in diuretic resistance. We propose a mechanism-based classification system of diuretic resistance by anatomical location as follows: pre-nephron resistance, pre-loop of Henle resistance, loop of Henle resistance, and post-loop of Henle resistance. Within this paradigm, we compare and contrast historical beliefs of resistance mechanisms with current literature specific to patients with heart failure. We recommend a treatment pathway to restore diuretic efficacy with a literature review of the various combination diuretic strategies and ongoing clinical trials that may impact current best practices.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Defining loop diuretic resistance in acute heart failure
Intravenous (IV) loop diuretic therapy is required in 80–90% of acute heart failure (AHF) hospitalizations to treat symptoms of hypervolemia[1, 2]. Quantifying the incidence of loop diuretic resistance is limited by the absence of a universal definition for this complication. Qualitatively, diuretic resistance is an unsatisfactory rate of diuresis/natriuresis despite an adequate diuretic regimen. This qualitative description consists of three subjective evaluations: (1) presence and magnitude of hypervolemia; (2) adequacy of the diuretic regimen; and (3) rate of net negative urine and sodium balance. Each component is interdependent, subjective to the evaluator, and problematic to measure (Table 1).
Diuretic response will decrease as euvolemia is approached, even if all other parameters remain constant. Ensuring the patient remains hypervolemic by the best available methods is the first step in defining diuretic resistance. Second, the adequacy of the diuretic dose and frequency must be addressed. Diuretic resistance is only considered when the loop diuretic regimen should yield diuresis, yet the rate of decongestion is inadequate. The determination of a diuretic regimen’s adequacy is subjective and may be evaluated relative to the oral outpatient dose, historical response, frequency, utilization of other concomitant diuretics, and kidney function. Although lacking a defined value for diuretic resistance, diuretic efficiency has significant prognostic implications [24]. Finally, the rate of decongestion must be assessed. Commonly used metrics such as weight changes and net input-output measurements are imprecise in clinical practice due to inaccurate measurements and other influential factors. Agreement between these two metrics is poor even in the setting of rigorous clinical trials (r = − 0.381 in the ASCEND-HF and r = 0.55 in DOSE-AHF clinical trials) [29, 30]. Furthermore, many AHF hospital admissions are not associated with weight gain, limiting the application of weight changes [31, 32].
Given these ambiguities, a quantitative definition of diuretic resistance with universal application remains elusive. Spot urinary sodium measurements are an emerging method to measure diuretic resistance. The spot urine sodium from a urine sample collected 1–2 h (h) after the administration of an IV loop diuretic dose can predict the total sodium excretion over the 6-h natriuretic duration of the loop diuretic with strong correlation with the measured 6-h sodium output (r = 0.91, p < 0.0001) by the following equation [25]:
Na = sodium; eGFR = estimated glomerular filtration rate, BSA = body surface area, CrSerum = serum creatinine; CrUrine = urine creatinine; NaUrine = urinary sodium concentration
Patients with a calculated cumulative sodium output < 100 mmol will not achieve a significantly negative sodium balance with twice daily diuretic dosing assuming a normal sodium restricted diet [25]. By identifying patients with natriuretic resistance within 1–2 h, clinicians can make rapid diuretic titrations to overcome diuretic resistance compared to traditional monitoring practices (Fig. 1). During consecutive days of diuresis, urinary sodium concentrations undergo significant fluctuation, can diminish despite preserved volumes of urine output, and may require serial measurement [3, 28].

Comparison of potential diuretic adjustment strategies. *Calculated 6-h total sodium output can be done using the equation in the text above, which is available as a free, online calculator at www.cardiorenalresearch.net. UOP, urine output; IV, intravenous; [UNa], spot urinary sodium concentration in millimole/liter; Total UNa, 6-h cumulative sodium output in millimole
Prognostic impact of diuretic resistance in acute heart failure
Diuretic resistance confers a worse prognosis, with the prognostic impact depending on the definition employed [33,34,35]. In the absence of randomized trials comparing therapies for diuretic resistance, it is difficult to discern the relative potential harm between the intertwined elements: diuretic resistance itself, the resultant increased loop diuretic doses, and the increased risk of not achieving decongestion. While the contribution of diuretic resistance is unknown, AHF hospitalizations ending without adequate decongestion are associated with worse outcomes and higher readmissions [36].
Loop diuretics increase neurohumoral activation regardless of the dose, diuretic response, or volume state [37, 38]. DOSE-AHF provides insight into the net balance between the decongestive benefits from higher diuretic doses and the potentially harmful neurohumoral activation, as it randomized patients to a high- or low-dose loop diuretic strategy [39]. The DOSE-AHF trial found no effect on 60-day death or rehospitalization between the high- or low-dose strategy, although the prevalence of diuretic resistance was unknown [40]. Patients randomized to a high-dose strategy had better 60-day outcomes, after adjusting for cumulative dose. However, the benefit was eliminated after adjusting for the resulting net urine output [39]. Changes in neurohormonal biomarkers during diuresis did not differ between the high- and low-dose groups and were not associated with 60-day outcomes in the DOSE-AHF trial [38]. While the potential for dose-related harm from loop diuretics cannot be excluded, the decongestive benefits of high-dose loop diuretics appear to offset potential harm.
Regarding the prognostic implications of diuretic resistance itself, a spot urine sodium less than 50–70 mmol/L after the first dose of IV loop diuretic is associated with higher risk of worsening kidney function, worsening heart failure (HF), and long-term adverse events [26, 41,42,43]. Yet, this metric does not consider the diuretic dose. Change in serial measures of spot urine sodium after diuretic adjustment may provide further prognostic value toward decongestive and long-term outcomes [44]. Diuretic efficiency is the best available metric to separate the prognostic effect of decongestive therapy intensity from resistance itself. Patients with diuretic efficiency below a population median had increased mortality (HR 3.57; 95% CI 1.46–8.73; p = 0.005), with those exhibiting low diuretic efficiency on high loop diuretic doses having the worst prognosis [24]. Consequently, diuretic resistance is known to confer a worse prognosis when high-dose loop diuretics are required with sustained low diuretic efficiency or resistance prohibits achievement of euvolemia with medical therapy.
Lastly, one must acknowledge that a mild resistance to diuretics can be beneficial. The term diuretic braking illustrates a beneficial adaptation to diuretics. Diuretic braking describes a diminished response to the same diuretic regimen [45]. If the initial diuretic response of excreting 20% of filtered sodium persisted, a continuous loop diuretic infusion would excrete 280 g of salt and 50 L of urine daily in a patient with an glomerular filtration rate (GFR) of 120 mL/min filtering 1400 g of sodium/day. In response to the immediate natriuresis, renal autoregulation and diuretic braking preserve the GFR. Diuretic braking is beneficial by ensuring loop diuretics do not have an unacceptably small therapeutic window. The term diuretic braking in clinical practice fails to characterize a specific mechanism of resistance or distinguish between beneficial renal adaptation and maladaptive diuretic resistance. Thus, a clinically actionable classification system should be employed instead.
Classification of loop diuretic resistance mechanisms
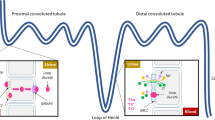
Diuretic resistance limiting decongestive goals, which may be similar or different mechanistically as beneficial diuretic braking, can be broadly categorized as pre-nephron diuretic resistance and intra-nephron diuretic resistance (Fig. 2). Intra-nephron diuretic resistance can further be divided into pre-loop of Henle diuretic resistance, loop of Henle diuretic resistance, and post-loop of Henle diuretic resistance. When evaluating diuretic resistance mechanism literature, one must consider the population studied. Many of the historical studies of diuretic resistance were performed in healthy controls or patients with hypertension or chronic kidney disease. The presumption that these findings can be intuitively applied to the AHF patient on modern medical therapies is flawed and has been challenged by recent literature specific to patients with HF.

Classification of potential IV loop diuretic resistance mechanisms. *Adapted with permission from Cardiorenal Syndrome in Heart Failure [46]
Pre-nephron diuretic resistance
Historical diuretic studies focused on pre-nephron and pre-loop of Henle resistance mechanisms in healthy subjects and patients with hypertension or chronic kidney disease [47,48,49,50,51,52,53]. Low cardiac output to the kidney, once thought to be a predominant driver of cardiorenal syndrome and diuretic resistance, has been proven by multiple recent analyses to be of minimal importance at the AHF population level [8, 9, 54]. Venous congestion, hypothesized to initiate diuretic resistance through a reduction in the arterial to venous pressure gradient at the glomerulus, was also unrelated to diuretic efficiency [24]. Vasodilators, dopamine, and milrinone failed to augment diuresis or weight loss in patients with AHF [55,56,57,58,59,60]. While dopamine trended toward increasing urine volume in those with a baseline systolic blood pressure (BP) less than 114 mmHg [57], antagonism of the renin-angiotensin-aldosterone system (RAAS) may improve natriuresis even in the setting of BP reduction [61, 62]. Activation of the RAAS varies significantly during decongestion and lacks association with diuretic dose or diuretic response, although the timing of RAAS biomarkers during decongestive therapy limits definitive conclusions [38, 63]. It remains unclear which patients with lower BP and diuretic resistance should have a temporary cessation in medications that lower BP versus those in whom RAAS antagonists should be continued or increased. Lastly, non-steroidal anti-inflammatory drugs should be discontinued, as they impair renal blood flow and natriuresis by inhibiting prostaglandin synthesis [64, 65].
Hypoalbuminemia has been investigated as a pre-nephron diuretic resistance mechanism because all loop diuretics are > 90% bound to albumin [66,67,68]. Hypothesized mechanisms include a reduced intravascular volume available for diuresis and decreased delivery of loop diuretics to the nephron [69]. The majority of literature evaluating the benefit of IV albumin replacement with IV furosemide was performed in nephrotic syndrome or cirrhosis utilizing IV furosemide doses of only 40 mg [50, 70, 71]. In a cohort of patients with HF and a medium serum albumin of 3.70 g/dL (IQR 3.50 to 4.10), serum albumin had no correlation with urinary diuretic delivery nor diuretic resistance measured as diuretic efficiency after adjustment for inflammatory markers [72]. Recent AHF trials have validated these results, finding no association between baseline serum albumin concentrations and weight loss (p = 0.43), diuretic efficiency (p = 0.53), or freedom from congestion (p = 0.30) [73].
The relationship between sodium and heart failure outcomes is complex, with insufficient evidence to recommend any specific dietary sodium intake for patients with AHF undergoing diuresis [74, 75]. Traditional paradigms consider high sodium intake to be a cause of pre-nephron diuretic resistance [47, 76]. In contrast, higher sodium intake might be beneficial in AHF populations if a greater net sodium removal is achieved [74]. Co-therapy with hypertonic saline and high-dose loop diuretics produced greater natriuresis and urine volume than high-dose loop diuretics alone among AHF patients with diuretic resistance [77,78,79]. However, the quality of data supporting this approach is limited. Hypertonic saline therapy cannot be recommended presently until the safety and efficacy is demonstrated in a larger, diverse population achieving a net negative sodium balance [80].
Pre-loop of Henle diuretic resistance
Kidney function and albuminuria, which are hypothesized to impair diuretic delivery to the site of action, are less influential mechanisms of diuretic resistance compared to tubular handling of sodium in HF. Animal models of nephrotic syndrome [81,82,83] indicated albuminuria caused diuretic resistance by binding loop diuretics in the urine as in the serum [84]. A recent study in humans with nephrotic syndrome has disproven albuminuria as a primary mechanism of diuretic resistance [85]. Patients with AHF and normal albuminuria (43%), microalbuminura (39%), or macroalbuminuria (18%) exhibited no correlation between diuretic efficiency and urinary albumin concentrations (r = − 0.145, p = 0.08) [72].
In the novel “The House of God,” we see renally based diuretic adjustments taught as “age + BUN = Lasix dose” [86], which contemporary medical pocket resources continue [84]. Unlike chronic kidney disease populations, renal dysfunction is less relevant in HF as a cause of diuretic resistance and is responsive to increased diuretic dose. Estimated glomerular filtration rate (eGFR) poorly correlates with net fluid output (r2 = 0.0; p = 0.35) and diuretic efficiency (r2 = 0.02; p < 0.001) in patients with AHF [24]. Elevated BUN but not reduced eGFR predicted urine output in the ASCEND-HF trial, which could reflect neurohumoral activation and/or reduced diuretic delivery to the site of action [55]. A cohort of patients with HF were studied to evaluate the relative importance of diuretic delivery and renal tubular response in diuretic resistance [87]. Urea clearance (r = 0.75; p = 0.001) and low eGFR (r = 0.58; p = 0.001) strongly correlated with decreased diuretic delivery to the kidney, but interestingly, patients with lower eGFR compensated for decreased diuretic concentrations by producing approximately 2-fold greater fractional excretion of sodium at 6 h. Kidney function in HF has no impact on the individual nephron’s net filtrate, but does influence total natriuresis through a reduction in the total number of nephrons. In summary, kidney dysfunction is much less of an important mediator of diuretic resistance in AHF than loop of Henle and post-loop of Henle diuretic resistance.
The proximal tubule is responsible for reabsorbing approximately 60% of filtered sodium [3]. Decreases in renal blood flow and increases in renal lymphatic flow secondary to HF may increase the percentage of filtered sodium reabsorbed up to 75% [88]. Further research is needed to quantify the contribution of the proximal convoluted tubule to diuretic resistance relative to resistance in the loop of Henle and distal tubules, although current literature indicates post-loop of Henle resistance is of greater significance.
Loop of Henle diuretic resistance
Loop diuretic’s dose-response curve exhibits a sigmoidal pattern along a logarithmic scale, with both a threshold and ceiling effect. The diuretic concentration in the lumen of the loop of Henle relative to the diuretic threshold determines the peak rate and duration of diuresis (Fig. 3). A dose exceeding the ceiling can still cause a greater diuretic response by maintaining a concentration above the threshold for a longer time. An IV loop diuretic dose that fails to cross the diuretic threshold will result in diuretic resistance. Likewise, a diuretic dose that elicits an adequate response can fail to meet the decongestive goals for the day if it is given with an inadequate frequency. Following a dose, urinary concentrations of loop diuretics fall below the diuretic threshold quickly (half-life 1–2 h) with a duration of action that rarely exceeds 6 h [67, 68]. Typical twice daily dosing may provide diuretic concentration below the diuretic threshold for the majority of the day, allowing compensatory sodium reabsorption [45, 89].

Loop diuretic pharmacokinetics and dose-response curve. The loop diuretic plasma concentration (y-axis) is plotted over time (x-axis) when given as an intravenous bolus. The Diuretic Threshold (red dotted line) is the diuretic concentration that must be exceeded to cause diuresis. The Diuretic Ceiling (blue dotted line) is the diuretic concentration above which no further increases in diuretic response are gained. The shaded area illustrates the area of the curve between the Diuretic Threshold and Ceiling. The boxed graph within the graph on the right shows the simultaneous sigmoidal dose-response relationship between the diuretic concentration (x-axis) and the natriuretic response (y-axis). The orange circle represents the moment the diuretic concentration crosses the Diuretic Threshold simultaneously in both graphs. The green circle represents the when the diuretic concentration reaches the Diuretic Ceiling simultaneously in both graphs
Post-loop of Henle diuretic resistance
Continuous loop diuretic exposure in animals has shown rapid distal tubular hypertrophy and hyperfunction [90,91,92]. The few contemporary studies in AHF patients indicate that the majority of diuretic resistance is primarily mediated by post-loop of Henle diuretic resistance. In patients with AHF, a median dose of IV furosemide 160 mg (40–270 mg) increased the amount of sodium estimated to exit the loop of Henle by 12.6 ± 10.8% (p < 0.001) compared to a pre-diuretic baseline [93]. The net fractional excretion of sodium only increased 4.8 ± 3.3%, indicating 66% (25–85%) of the sodium leaving the loop of Henle underwent distal tubular reabsorption. The authors controlled for loop of Henle diuretic resistance by using urine diuretic concentration and reported the increase in sodium leaving the loop of Henle only accounted for 6.4% of the increase in net fractional excretion of sodium. A separate study of AHF patients receiving IV loop diuretic corroborated these findings, quantifying the majority (71%) of diuretic response was related to intra-renal diuretic resistance via renal tubular changes [87].
Diuretic strategies to overcome diuretic resistance
The following discussion assumes the patient exhibiting diuretic resistance is hemodynamically stable and hypervolemic. Additionally, it assumes the clinician has excluded causes of pseudo-resistance, such as drug interactions (NSAIDs, probenecid), urinary tract obstruction, or total body euvolemia with edema secondary to lymphedema or hypoalbuminemia. Medical therapy should always be individualized to the diuretic resistance mechanism when known to restore diuretic efficacy and achieve clinical euvolemia. A stepwise approach to diuretic titration based upon diuretic response with prioritization of loop diuretic optimization was employed in CARRESS-HF and represents the current best practice of overcoming diuretic resistance [3, 94]. We propose the following approach to the patient with AHF and diuretic resistance based upon the relative incidence of diuretic resistance mechanisms and the current literature supporting efficacy and safety of the diuretic therapies.
-
1.
Address loop of Henle resistance mechanisms
Diuretic dose and frequency are interdependent in loop of Henle diuretic resistance and both must be considered. When evaluating for loop of Henle diuretic resistance, the natriuretic response to the dose should first be considered. A spot urine sodium less than 50 to 70 mmol/L or a urine output rate less than 600 mL over 6 h necessitates an increase in the loop diuretic dose [3] (Fig. 1). If the spot urine sodium is > 70 mmol/L, calculation of the 6-h cumulative urine sodium output provides additional guidance. Using the 6-h cumulative sodium output, clinicians can modify the loop diuretic regimen’s dose and/or frequency to produce a net negative sodium balance relative to the daily dietary sodium intake (2 g sodium diet = 87 mmol). If an adequate natriuretic response is achieved, the frequency should be addressed next (Fig. 4). Continuous infusions of loop diuretics should be advantageous by consistently exceeding the diuretic threshold [95]. Yet, the DOSE-AHF trial found no difference in symptom improvement, urine output, or weight loss when administering the same total loop diuretic daily dose divided twice daily versus a continuous infusion in patients with an unknown prevalence of diuretic resistance [40]. In patients exhibiting diuretic resistance but with adequate natriuretic response to an IV bolus dose, consideration can be given to the use of IV loop diuretics at greater frequencies to overcome frequency-mediated diuretic resistance, although data proving this theoretical approach is lacking. This strategy differs from the DOSE-AHF trials’ null findings in that more frequent dosing also represents an increase of the total daily diuretic dose in addition to increased frequency.
-
2.
Address post-loop of Henle diuretic resistance

Diuretic strategies to overcome diuretic resistance
Although post-loop of Henle is the resistance mechanism in the majority of patients AHF, clinicians should first ensure an adequate loop diuretic dose and frequency are prescribed before employing combination nephron blockade targeting post-loop of Henle diuretic resistance (Fig. 4). Combination nephron blockade with metolazone was investigated in an observational cohort of 13,898 AHF hospital admissions, of which 1048 utilized adjuvant metolazone [96]. After propensity and covariate adjusted analyses, adding metolazone was associated with increased risk of hypokalemia (OR 2.80; 95% CI 2.25–3.50), hyponatremia (OR 2.13; 95% CI 1.73–2.62), worsening renal function (OR 3.02; 95% CI 2.55–3.58), and mortality (OR 1.20; 95% CI 1.04–1.39) [96]. In contrast, the use of high-dose loop diuretics did not have any association with harm. Interestingly, the harm associated with metolazone was only in patients who did not have metolazone added to high-dose loop diuretics. Together with the DOSE-AHF trial’s absence of harm between high and low dose diuretics, the limited current literature indicates escalation of loop diuretic doses may be the preferred method until randomized, comparative trials (NCT01647932) can better inform practice [3, 40, 97].
Thiazide (and thiazide-like) medications are the most commonly utilized medications to overcome post-loop of Henle diuretic resistance [98,99,100]. Since thiazides inhibit sodium reabsorption in the distal convoluted tubules where the majority of remaining sodium reabsorption occurs after the loop of Henle, these agents should be the initial agent chosen for combination nephron blockade. Despite experience spanning 50 years, common misconceptions regarding thiazides in combination nephron blockade persist [99]. Thiazides appear to have equal efficacy at equipotent doses [101]; therefore, decisions between agents should be based upon pharmacokinetic differences, particularly among agents with additional carbonic anhydrase antagonistic ability. Although metolazone is often considered superior to other thiazides, no solid evidence supports this perception, even in patients with low eGFR [99, 102,103,104]. Administration of thiazides 30 min prior to loop diuretics is not based upon evidence, as most studies administered both agents simultaneously [99]. The erratic and delayed absorption of metolazone makes this practice clinically irrelevant and unnecessarily increases complexity [105, 106].
While oral chlorothiazide is not utilized secondary to poor absorption, IV chlorothiazide offers the pharmacokinetic advantages of a quicker onset of action compared to metolazone’s slow absorption and has a shorter duration of action that may better facilitate titration to diuretic response [101]. To date, no randomized trials have compared IV chlorothiazide to oral thiazides, limiting definitive conclusions on efficacy differences [107]. Two ongoing randomized clinical trials (NCT02606253 and NCT03574857) comparing metolazone and IV chlorothiazide will provide further insight into this issue.
Careful monitoring for electrolyte abnormalities, kidney function, and volume status is warranted with all thiazides to avoid adverse events [99]. The risk of hypochloremia increases in combination nephron blockade and is emerging as a research target [96, 108]. Hypochloremia has been associated with increased mortality risk and diuretic resistance [109,110,111,112]. No data currently exists to guide chloride supplementation or modification of combination diuretic therapy on the basis of serum chloride.
Mineralocorticoid receptor antagonists and epithelial sodium channel inhibitors impact the late distal tubule and collecting duct. Given the reduced capacity for sodium reabsorption in this anatomical area compared to the site of action of thiazides, these agents are thought unlikely to provide superior diuretic effects in combination with loop diuretics [3, 113]. Diuretic doses of mineralocorticoid receptor antagonists are combined with loop diuretics in cirrhotic ascites as the primary combination nephron blockade strategy [114, 115]. In chronic HF, non-diuretic doses (< 50 mg/day) of mineralocorticoid receptor antagonists reduce morbidity and mortality [116]. Literature examining higher doses of mineralocorticoid receptor antagonists with the intent of augmenting diuresis is scarce [117, 118]. The ATHENA-HF trial compared spironolactone 100 mg/day to placebo or continued non-diuretic dose spironolactone (25 mg) in patients with hypervolemic AHF treated with IV loop diuretics [119]. No difference in the primary endpoint of natriuretic peptide change or secondary diuretic outcomes such as net urine output, weight change, or IV loop diuretic doses required were found [119]. Several factors should be considered when interpreting these results. The population studied did not exhibit diuretic resistance, receiving a median IV furosemide daily dose of 160 mg (IQR 100, 320). Spironolactone has a short half-life (1.5 h), and the active metabolite canrenone (half-life ~ 17 h) is responsible for the majority of the medication’s effects [120]. As steady-state canrenone concentrations are not achieved until day 3 of therapy, the 96-h time period might be insufficient to measure the effects [119]. Serum potassium levels were no different between spironolactone and placebo, supporting this possibility [119]. Future studies should investigate higher doses of spironolactone (200–400 mg/day) or other mineralocorticoid receptor antagonists in a diuretic resistance population. Currently in AHF with diuretic resistance, this class can be utilized for hypokalemia management and continued as a part of chronic neurohormonal therapies, but diuretic doses should be reserved until failure of combination nephron blockade with loops and thiazides.
Vasopressin-2 receptor antagonists have been extensively investigated in AHF. Earlier trials evaluated the impact on mortality and hyponatremia [121, 122]. Recent investigations have re-focused the primary efficacy endpoints to study their decongestive effects. Vasopressin-2 receptor antagonists exert diuretic effects by blocking vasopressin-mediated aquaporin channels in the collecting duct, causing an increase in urinary water (aquaresis) when used alone [123]. The natriuretic potential when combined with high-dose loop diuretics in patients with diuretic resistance is not described to date. Ongoing clinical trials are investigating the natriuretic effects with this application (NCT02606253), which will be critical if a urine sodium-based diuretic strategy is employed (Fig. 4). Three trials comparing tolvaptan to placebo in patients with hypervolemic AHF without diuretic resistance treated with only modest IV loop diuretics (mean daily IV furosemide equivalent 80–160 mg) found increases in weight loss and urine output [124,125,126]. Tolvaptan cannot be recommended over thiazides in combination nephron blockage at this time given the limited study in loop diuretic resistance.
The proximal convoluted tubule reabsorbs the largest percentage of filtered sodium, making it an attractive target for diuretic therapies [113]. Medications acting in the proximal convoluted tubules with potential for combination nephron blockade include acetazolamide and sodium-glucose co-transporter 2 (SGLT2) inhibitors. Acetazolamide has a number of potentially positive extra-diuretic effects, including increased salt delivery to the macula densa reducing neurohormonal activation [127]. When combined with low doses of oral and IV furosemide in small cohorts of patients with HF but without diuretic resistance, acetazolamide increased the natriuretic response [128, 129]. Acetazolamide is currently being investigated in combination with IV loop diuretics in the ADVOR trial (NCT03505788) [127]. SGLT2 inhibitors have several ongoing clinical trials to establish the acute natriuretic effects in patients with HF, but their use as adjunctive diuretic agents cannot be recommended currently. Acetazolamide may be a promising diuretic to add when diuretic resistance persists despite combination nephron blockade with loop diuretics and thiazides, but there is no conclusive evidence to recommend its use over thiazides currently.
In conclusion, a universally applicable, quantitative definition of diuretic resistance in AHF remains elusive. The mechanisms behind diuretic resistance are diverse. A mechanism-based classification can guide medical strategies to restore diuretic efficacy. Optimization of loop diuretic regimens based upon diuretic response should be the primary strategy followed by combination nephron blockage with thiazides. Novel diuretic combination strategies are emerging but require further research before they can be recommended.
References
Fonarow GC, Heywood JT, Heidenreich PA, Lopatin M, Yancy CW (2007) Temporal trends in clinical characteristics, treatments, and outcomes for heart failure hospitalizations, 2002 to 2004: findings from Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J 153:1021–1028
Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, Nodari S, Lam CS, Sato N, Shah AN, Gheorghiade M (2014) The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol 63:1123–1133
Mullens W, Damman K, Harjola VP, Mebazaa A, Brunner-La Rocca HP, Martens P, Testani JM, Tang WHW, Orso F, Rossignol P, Metra M, Filippatos G, Seferovic PM, Ruschitzka F, Coats AJ (2019) The use of diuretics in heart failure with congestion—a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 21:137–155
Chakko S, Woska D, Martinez H, de Marchena E, Futterman L, Kessler KM, Myerberg RJ (1991) Clinical, radiographic, and hemodynamic correlations in chronic congestive heart failure: conflicting results may lead to inappropriate care. Am J Med 90:353–359
Elder A, Japp A, Verghese A (2016) How valuable is physical examination of the cardiovascular system? BMJ. 354:i3309
Drazner MH, Hellkamp AS, Leier CV, Shah MR, Miller LW, Russell SD, Young JB, Califf RM, Nohria A (2008) Value of clinician assessment of hemodynamics in advanced heart failure: the ESCAPE trial. Circ Heart Fail 1:170–177
Stevenson LW, Perloff JK (1989) The limited reliability of physical signs for estimating hemodynamics in chronic heart failure. JAMA 261:884–888
Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, Young JB, Tang WH (2009) Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 53:589–596
Nohria A, Hasselblad V, Stebbins A, Pauly DF, Fonarow GC, Shah M, Yancy CW, Califf RM, Stevenson LW, Hill JA (2008) Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol 51:1268–1274
Ronco C, Haapio M, House AA, Anavekar N, Bellomo R (2008) Cardiorenal syndrome. J Am Coll Cardiol 52:1527–1539
Damman K, Testani JM (2015) The kidney in heart failure: an update. Eur Heart J 36:1437–1444
Testani JM, Kimmel SE, Dries DL, Coca SG (2011) Prognostic importance of early worsening renal function after initiation of angiotensin-converting enzyme inhibitor therapy in patients with cardiac dysfunction. Circ Heart Fail 4:685–691
Michel A, Martin-Perez M, Ruigomez A, Garcia Rodriguez LA (2016) Incidence and risk factors for severe renal impairment after first diagnosis of heart failure: a cohort and nested case-control study in UK general practice. Int J Cardiol 207:252–257
Daniels LB, Maisel AS (2007) Natriuretic peptides. J Am Coll Cardiol 50:2357–2368
Desai AS (2013) Are serial BNP measurements useful in heart failure management? Serial natriuretic peptide measurements are not useful in heart failure management: the art of medicine remains long. Circulation. 127:509–516 discussion 516
Stienen S, Salah K, Moons AH, Bakx AL, van Pol P, Kortz RAM, Ferreira JP, Marques I, Schroeder-Tanka JM, Keijer JT, Bayes-Genis A, Tijssen JGP, Pinto YM, Kok WE (2018) NT-proBNP (N-terminal pro-B-type natriuretic peptide)-guided therapy in acute decompensated heart failure: PRIMA II randomized controlled trial (can NT-ProBNP-guided therapy during hospital admission for acute decompensated heart failure reduce mortality and readmissions?). Circulation. 137:1671–1683
Kociol RD, McNulty SE, Hernandez AF, Lee KL, Redfield MM, Tracy RP, Braunwald E, O’Connor CM, Felker GM, NHLBI Heart Failure Network Steering Committee and Investigators (2013) Markers of decongestion, dyspnea relief, and clinical outcomes among patients hospitalized with acute heart failure. Circ Heart Fail 6:240–245
Cooper LB, Mentz RJ, Gallup D, Lala A, DeVore AD, Vader JM, AbouEzzeddine OF, Bart BA, Anstrom KJ, Hernandez AF, Felker GM (2016) Serum bicarbonate in acute heart failure: relationship to treatment strategies and clinical outcomes. J Card Fail 22:738–742
Lala A, McNulty SE, Mentz RJ, Dunlay SM, Vader JM, AbouEzzeddine OF, DeVore AD, Khazanie P, Redfield MM, Goldsmith SR, Bart BA, Anstrom KJ, Felker GM, Hernandez AF, Stevenson LW (2015) Relief and recurrence of congestion during and after hospitalization for acute heart failure: insights from Diuretic Optimization Strategy Evaluation in Acute Decompensated Heart Failure (DOSE-AHF) and Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARESS-HF). Circ Heart Fail 8:741–748
Miller WL (2016) Fluid volume overload and congestion in heart failure: time to reconsider pathophysiology and how volume is assessed. Circ Heart Fail 9:e002922
Neuberg GW, Miller AB, O’Connor CM, Belkin RN, Carson PE, Cropp AB, Frid DJ, Nye RG, Pressler ML, Wertheimer JH, Packer M (2002) Diuretic resistance predicts mortality in patients with advanced heart failure. Am Heart J 144:31–38
Testani JM, Cappola TP, Brensinger CM, Shannon RP, Kimmel SE (2011) Interaction between loop diuretic-associated mortality and blood urea nitrogen concentration in chronic heart failure. J Am Coll Cardiol 58:375–382
Yilmaz MB, Gayat E, Salem R, Lassus J, Nikolaou M, Laribi S, Parissis J, Follath F, Peacock WF, Mebazaa A (2011) Impact of diuretic dosing on mortality in acute heart failure using a propensity-matched analysis. Eur J Heart Fail 13:1244–1252
Testani JM, Brisco MA, Turner JM, Spatz ES, Bellumkonda L, Parikh CR, Tang WH (2014) Loop diuretic efficiency: a metric of diuretic responsiveness with prognostic importance in acute decompensated heart failure. Circ Heart Fail 7:261–270
Testani JM, Hanberg JS, Cheng S, Rao V, Onyebeke C, Laur O, Kula A, Chen M, Wilson FP, Darlington A, Bellumkonda L, Jacoby D, Tang WH, Parikh CR (2016) Rapid and highly accurate prediction of poor loop diuretic natriuretic response in patients with heart failure. Circ Heart Fail 9:e002370
Brinkley DM Jr, Burpee LJ, Chaudhry SP, Smallwood JA, Lindenfeld J, Lakdawala NK, Desai AS, Stevenson LW (2018) Spot urine sodium as triage for effective diuretic infusion in an ambulatory heart failure unit. J Card Fail 24:349–354
Singh D, Shrestha K, Testani JM, Verbrugge FH, Dupont M, Mullens W, Tang WH (2014) Insufficient natriuretic response to continuous intravenous furosemide is associated with poor long-term outcomes in acute decompensated heart failure. J Card Fail 20:392–399
Verbrugge FH, Nijst P, Dupont M, Penders J, Tang WH, Mullens W (2014) Urinary composition during decongestive treatment in heart failure with reduced ejection fraction. Circ Heart Fail 7:766–772
Ambrosy AP, Cerbin LP, Armstrong PW, Butler J, Coles A, DeVore AD, Dunlap ME, Ezekowitz JA, Felker GM, Fudim M, Greene SJ, Hernandez AF, O’Connor CM, Schulte P, Starling RC, Teerlink JR, Voors AA, Mentz RJ (2017) Body weight change during and after hospitalization for acute heart failure: patient characteristics, markers of congestion, and outcomes: findings from the ASCEND-HF trial. JACC Heart Fail 5:1–13
Testani JM, Brisco MA, Kociol RD, Jacoby D, Bellumkonda L, Parikh CR, Coca SG, Tang WH (2015) Substantial discrepancy between fluid and weight loss during acute decompensated heart failure treatment. Am J Med 128:776–783 e4
Zile MR, Bennett TD, St John Sutton M, Cho YK, Adamson PB, Aaron MF, Aranda JM Jr, Abraham WT, Smart FW, Stevenson LW, Kueffer FJ, Bourge RC (2008) Transition from chronic compensated to acute decompensated heart failure: pathophysiological insights obtained from continuous monitoring of intracardiac pressures. Circulation. 118:1433–1441
Chaudhry SI, Wang Y, Concato J, Gill TM, Krumholz HM (2007) Patterns of weight change preceding hospitalization for heart failure. Circulation. 116:1549–1554
Patarroyo M, Wehbe E, Hanna M, Taylor DO, Starling RC, Demirjian S, Tang WH (2012) Cardiorenal outcomes after slow continuous ultrafiltration therapy in refractory patients with advanced decompensated heart failure. J Am Coll Cardiol 60:1906–1912
Kiernan MS, Stevens SR, Tang WHW, Butler J, Anstrom KJ, Birati EY, Grodin JL, Gupta D, Margulies KB, LaRue S, Davila-Roman VG, Hernandez AF, de Las FL, Investigators NHFCTN (2018) Determinants of diuretic responsiveness and associated outcomes during acute heart failure hospitalization: an analysis from the NHLBI heart failure network clinical trials. J Card Fail 24:428–438
Valente MA, Voors AA, Damman K, Van Veldhuisen DJ, Massie BM, O’Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, Davison B, Cleland JG, Givertz MM, Bloomfield DM, Fiuzat M, Dittrich HC, Hillege HL (2014) Diuretic response in acute heart failure: clinical characteristics and prognostic significance. Eur Heart J 35:1284–1293
Rubio-Gracia J, Demissei BG, Ter Maaten JM, Cleland JG, O’Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, Davison BA, Givertz MM, Bloomfield DM, Dittrich H, Damman K, Perez-Calvo JI, Voors AA (2018) Prevalence, predictors and clinical outcome of residual congestion in acute decompensated heart failure. Int J Cardiol 258:185–191
Felker GM, Mentz RJ (2012) Diuretics and ultrafiltration in acute decompensated heart failure. J Am Coll Cardiol 59:2145–2153
Mentz RJ, Stevens SR, DeVore AD, Lala A, Vader JM, AbouEzzeddine OF, Khazanie P, Redfield MM, Stevenson LW, O’Connor CM, Goldsmith SR, Bart BA, Anstrom KJ, Hernandez AF, Braunwald E, Felker GM (2015) Decongestion strategies and renin-angiotensin-aldosterone system activation in acute heart failure. JACC Heart Fail 3:97–107
Hanberg JS, Tang WHW, Wilson FP, Coca SG, Ahmad T, Brisco MA, Testani JM (2017) An exploratory analysis of the competing effects of aggressive decongestion and high-dose loop diuretic therapy in the DOSE trial. Int J Cardiol 241:277–282
Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter MM, Deswal A, Rouleau JL, Ofili EO, Anstrom KJ, Hernandez AF, McNulty SE, Velazquez EJ, Kfoury AG, Chen HH, Givertz MM, Semigran MJ, Bart BA, Mascette AM, Braunwald E, O’Connor CM (2011) Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 364:797–805
Honda S, Nagai T, Nishimura K, Nakai M, Honda Y, Nakano H, Iwakami N, Sugano Y, Asaumi Y, Aiba T, Noguchi T, Kusano K, Yokoyama H, Ogawa H, Yasuda S, Anzai T, NaDEF Investigators (2018) Long-term prognostic significance of urinary sodium concentration in patients with acute heart failure. Int J Cardiol 254:189–194
Collins SP, Jenkins CA, Baughman A, Miller KF, Storrow AB, Han JH, Brown NJ, Liu D, Luther JM, McNaughton CD, Self WH, Peng D, Testani JM, Lindenfeld J (2019) Early urine electrolyte patterns in patients with acute heart failure. ESC Heart Fail 6:80–88
Luk A, Groarke JD, Desai AS, Mahmood SS, Gopal DM, Joyce E, Shah SP, Lindenfeld J, Stevenson L, Lakdawala NK (2018) First spot urine sodium after initial diuretic identifies patients at high risk for adverse outcome after heart failure hospitalization. Am Heart J 203:95–100
Biegus J, Zymlinski R, Sokolski M, Todd J, Cotter G, Metra M, Jankowska EA, Banasiak W, Ponikowski P (2019) Serial assessment of spot urine sodium predicts effectiveness of decongestion and outcome in patients with acute heart failure. Eur J Heart Fail 21:624–633
Ellison DH, Felker GM (2017) Diuretic treatment in heart failure. N Engl J Med 377:1964–1975
Cox ZL, Testani JM (2020) Loop diuretic resistance in a patient with acute heart failure. In: Tang W, Verbrugge F, Mullens W (eds) Cardiorenal syndrome in heart failure. Springer, Cham. https://doi.org/10.1007/978-3-030-21033-5_11
Brater DC (1998) Diuretic therapy. N Engl J Med 339:387–395
Wilcox CS (2002) New insights into diuretic use in patients with chronic renal disease. J Am Soc Nephrol 13:798–805
Beermann B, Midskov C (1986) Reduced bioavailability and effect of furosemide given with food. Eur J Clin Pharmacol 29:725–727
Phakdeekitcharoen B, Boonyawat K (2012) The added-up albumin enhances the diuretic effect of furosemide in patients with hypoalbuminemic chronic kidney disease: a randomized controlled study. BMC Nephrol 13:92
Loon NR, Wilcox CS, Unwin RJ (1989) Mechanism of impaired natriuretic response to furosemide during prolonged therapy. Kidney Int 36:682–689
McCrindle JL, Li Kam Wa TC, Barron W, Prescott LF (1996) Effect of food on the absorption of furosemide and bumetanide in man. Br J Clin Pharmacol 42:743–746
Nakahama H, Orita Y, Yamazaki M, Itoh S, Okuda T, Yamaji A, Miwa Y, Yanase M, Fukuhara Y, Kamada T (1988) Pharmacokinetic and pharmacodynamic interactions between furosemide and hydrochlorothiazide in nephrotic patients. Nephron. 49:223–227
Bock JS, Gottlieb SS (2010) Cardiorenal syndrome: new perspectives. Circulation. 121:2592–2600
Gottlieb SS, Stebbins A, Voors AA, Hasselblad V, Ezekowitz JA, Califf RM, O’Connor CM, Starling RC, Hernandez AF (2013) Effects of nesiritide and predictors of urine output in acute decompensated heart failure: results from ASCEND-HF. J Am Coll Cardiol 62:1177–1183
Cuffe MS, Califf RM, Adams KF Jr, Benza R, Bourge R, Colucci WS, Massie BM, O’Connor CM, Pina I, Quigg R, Silver MA, Gheorghiade M (2002) Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA 287:1541–1547
Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigram MJ, Goldsmith SR, Bart BA, Bull DA, Stehlik J, LeWinter MM, Konstam MA, Huggins GS, Rouleau JL, O’Meara E, Tang WH, Starling RC, Butler J, Deswal A, Felker GM, O’Connor CM, Bonita RE, Margulies KB, Cappola TP, Ofili EO, Mann DL, Davila-Roman VG, McNulty SE, Borlaug BA, Velazquez EJ, Lee KL, Shah MR, Hernandez AF, Braunwald E, Redfield MM (2013) Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 310:2533–2543
Packer M, O’Connor C, McMurray JJV, Wittes J, Abraham WT, Anker SD, Dickstein K, Filippatos G, Holcomb R, Krum H, Maggioni AP, Mebazaa A, Peacock WF, Petrie MC, Ponikowski P, Ruschitzka F, van Veldhuisen DJ, Kowarski LS, Schactman M, Holzmeister J, True-AHF Investigators (2017) Effect of ularitide on cardiovascular mortality in acute heart failure. N Engl J Med 376:1956–1964
Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr, Dorobantu MI, Grinfeld LR, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin TM, Metra M, RELAX-AHF Investigators (2013) Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 381:29–39
Sharma K, Vaishnav J, Kalathiya R, Hu JR, Miller J, Shah N, Hill T, Sharp M, Tsao A, Alexander KM, Gupta R, Montemayor K, Kovell L, Chasler JE, Lee YJ, Fine DM, Kass DA, Weiss RG, Thiemann DR, Ndumele CE, Schulman SP, Russell SD, Osler Medical Housestaff (2018) Randomized evaluation of heart failure with preserved ejection fraction patients with acute heart failure and dopamine: the ROPA-DOP trial. JACC Heart Fail 6:859–870
Chen HH, Redfield MM, Nordstrom LJ, Cataliotti A, Burnett JC Jr (2003) Angiotensin II AT1 receptor antagonism prevents detrimental renal actions of acute diuretic therapy in human heart failure. Am J Physiol Renal Physiol 284:F1115–F1119
Kula AJ, Hanberg JS, Wilson FP, Brisco MA, Bellumkonda L, Jacoby D, Coca SG, Parikh CR, Tang WHW, Testani JM (2016) Influence of titration of neurohormonal antagonists and blood pressure reduction on renal function and decongestion in decompensated heart failure. Circ Heart Fail 9:e002333
Guazzi MD, Agostoni P, Perego B, Lauri G, Salvioni A, Giraldi F, Matturri M, Guazzi M, Marenzi G (1994) Apparent paradox of neurohumoral axis inhibition after body fluid volume depletion in patients with chronic congestive heart failure and water retention. Br Heart J 72:534–539
Wilson DR, Honrath U, Sonnenberg H (1983) Furosemide action on collecting ducts: effect of prostaglandin synthesis inhibition. Am J Phys 244:F666–F673
Nies AS, Gal J, Fadul S, Gerber JG (1983) Indomethacin-furosemide interaction: the importance of renal blood flow. J Pharmacol Exp Ther 226:27–32
Torsemide [package insert]. Roche Pharmaceuticals IN, NJ.; April 2003
Furosemide [package insert]. Sanofi-aventis L, Bridgewater, NJ; November 2012
Bumetanide [package insert]. Bedford Pharmaceuticals I, Bedford, OH.; February 2010
Arques S, Ambrosi P (2011) Human serum albumin in the clinical syndrome of heart failure. J Card Fail 17:451–458
Chalasani N, Gorski JC, Horlander JC Sr, Craven R, Hoen H, Maya J, Brater DC (2001) Effects of albumin/furosemide mixtures on responses to furosemide in hypoalbuminemic patients. J Am Soc Nephrol 12:1010–1016
Elwell RJ, Spencer AP, Eisele G (2003) Combined furosemide and human albumin treatment for diuretic-resistant edema. Ann Pharmacother 37:695–700
Charokopos A, Sury K, Rao V, Ahmad T, Griffin M, Turner JM, Mahoney D, Cox ZL, Wilson FP and Testani JM (2019) The Influence of Serum and Urine Albumin on Loop Diuretic Response in Heart Failure. Clin J Am Soc Nephrol 14 (5) 712–718. https://doi.org/10.2215/CJN.11600918
Grodin JL, Lala A, Stevens SR, DeVore AD, Cooper LB, AbouEzzeddine OF, Mentz RJ, Groarke JD, Joyce E, Rosenthal JL, Vader JM, Tang WH (2016) Clinical implications of serum albumin levels in acute heart failure: insights from DOSE-AHF and ROSE-AHF. J Card Fail 22:884–890
Gupta D, Georgiopoulou VV, Kalogeropoulos AP, Dunbar SB, Reilly CM, Sands JM, Fonarow GC, Jessup M, Gheorghiade M, Yancy C, Butler J (2012) Dietary sodium intake in heart failure. Circulation. 126:479–485
Nijst P, Verbrugge FH, Grieten L, Dupont M, Steels P, Tang WHW, Mullens W (2015) The pathophysiological role of interstitial sodium in heart failure. J Am Coll Cardiol 65:378–388
Ellison DH (2001) Diuretic therapy and resistance in congestive heart failure. Cardiology. 96:132–143
Paterna S, Di Pasquale P, Parrinello G, Amato P, Cardinale A, Follone G, Giubilato A, Licata G (2000) Effects of high-dose furosemide and small-volume hypertonic saline solution infusion in comparison with a high dose of furosemide as a bolus, in refractory congestive heart failure. Eur J Heart Fail 2:305–313
Paterna S, Di Pasquale P, Parrinello G, Fornaciari E, Di Gaudio F, Fasullo S, Giammanco M, Sarullo FM, Licata G (2005) Changes in brain natriuretic peptide levels and bioelectrical impedance measurements after treatment with high-dose furosemide and hypertonic saline solution versus high-dose furosemide alone in refractory congestive heart failure: a double-blind study. J Am Coll Cardiol 45:1997–2003
Licata G, Di Pasquale P, Parrinello G, Cardinale A, Scandurra A, Follone G, Argano C, Tuttolomondo A, Paterna S (2003) Effects of high-dose furosemide and small-volume hypertonic saline solution infusion in comparison with a high dose of furosemide as bolus in refractory congestive heart failure: long-term effects. Am Heart J 145:459–466
Gandhi S, Mosleh W, Myers RB (2014) Hypertonic saline with furosemide for the treatment of acute congestive heart failure: a systematic review and meta-analysis. Int J Cardiol 173:139–145
Kirchner KA, Voelker JR, Brater DC (1990) Intratubular albumin blunts the response to furosemide—a mechanism for diuretic resistance in the nephrotic syndrome. J Pharmacol Exp Ther 252:1097–1101
Kirchner KA, Voelker JR, Brater DC (1991) Binding inhibitors restore furosemide potency in tubule fluid containing albumin. Kidney Int 40:418–424
Green TP, Mirkin BL (1980) Resistance of proteinuric rats to furosemide: urinary drug protein binding as a determinant of drug effect. Life Sci 26:623–630
Sabatine MS, Massachusetts General Hospital (2017) Pocket medicine: The Massachusetts General Hospital Handbook of Internal medicine. Sixth edition. Wolters Kluwer
Agarwal R, Gorski JC, Sundblad K, Brater DC (2000) Urinary protein binding does not affect response to furosemide in patients with nephrotic syndrome. J Am Soc Nephrol 11:1100–1105
Shem S (1979) The house of god. Bodley Head, London
Ter Maaten JM, Rao VS, Hanberg JS, Perry Wilson F, Bellumkonda L, Assefa M, Sam Broughton J, D'Ambrosi J, Wilson Tang WH, Damman K, Voors AA, Ellison DH, Testani JM (2017) Renal tubular resistance is the primary driver for loop diuretic resistance in acute heart failure. Eur J Heart Fail 19:1014–1022
Mullens W, Verbrugge FH, Nijst P, Tang WHW (2017) Renal sodium avidity in heart failure: from pathophysiology to treatment strategies. Eur Heart J 38:1872–1882
Cox ZL, Lenihan DJ (2014) Loop diuretic resistance in heart failure: resistance etiology-based strategies to restoring diuretic efficacy. J Card Fail 20:611–622
Ellison DH (1991) The physiologic basis of diuretic synergism: its role in treating diuretic resistance. Ann Intern Med 114:886–894
Stanton BA, Kaissling B (1988) Adaptation of distal tubule and collecting duct to increased Na delivery. II. Na+ and K+ transport. Am J Phys 255:F1269–F1275
Stanton BA, Kaissling B (1989) Regulation of renal ion transport and cell growth by sodium. Am J Phys 257:F1–F10
Rao VS, Planavsky N, Hanberg JS, Ahmad T, Brisco-Bacik MA, Wilson FP, Jacoby D, Chen M, Tang WHW, Cherney DZI, Ellison DH, Testani JM (2017) Compensatory distal reabsorption drives diuretic resistance in human heart failure. J Am Soc Nephrol 28:3414–3424
Bart BA, Goldsmith SR, Lee KL, Redfield MM, Felker GM, O’Connor CM, Chen HH, Rouleau JL, Givertz MM, Semigran MJ, Mann D, Deswal A, Bull DA, Lewinter MM, Braunwald E (2012) Cardiorenal rescue study in acute decompensated heart failure: rationale and design of CARRESS-HF, for the Heart Failure Clinical Research Network. J Card Fail 18:176–182
Felker GM, O’Connor CM, Braunwald E (2009) Loop diuretics in acute decompensated heart failure: necessary? Evil? A necessary evil? Circ Heart Fail 2:56–62
Brisco-Bacik MA, Ter Maaten JM, Houser SR, Vedage NA, Rao V, Ahmad T, Wilson FP, Testani JM (2018) Outcomes associated with a strategy of adjuvant metolazone or high-dose loop diuretics in acute decompensated heart failure: a propensity analysis. J Am Heart Assoc 7:e009149
Trullas JC, Morales-Rull JL, Casado J, Freitas Ramirez A, Manzano L, Formiga F, CLOROTIC Investigators (2016) Rationale and design of the “Safety and efficacy of the combination of loop with thiazide-type diuretics in patients with decompensated heart failure (CLOROTIC) trial:” a double-blind, randomized, placebo-controlled study to determine the effect of combined diuretic therapy (loop diuretics with thiazide-type diuretics) among patients with decompensated heart failure. J Card Fail 22:529–536
Bart BA, Goldsmith SR, Lee KL, Givertz MM, O’Connor CM, Bull DA, Redfield MM, Deswal A, Rouleau JL, LeWinter MM, Ofili EO, Stevenson LW, Semigran MJ, Felker GM, Chen HH, Hernandez AF, Anstrom KJ, McNulty SE, Velazquez EJ, Ibarra JC, Mascette AM, Braunwald E (2012) Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 367:2296–2304
Jentzer JC, DeWald TA, Hernandez AF (2010) Combination of loop diuretics with thiazide-type diuretics in heart failure. J Am Coll Cardiol 56:1527–1534
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr., Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ and Wilkoff BL. (2013) ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 128:e240–e327
Goodman LS, Brunton LL, Chabner B, Knollmann BC (2011) Goodman & Gilman’s the pharmacological basis of therapeutics, 12th edn. McGraw-Hill, New York
Channer KS, McLean KA, Lawson-Matthew P, Richardson M (1994) Combination diuretic treatment in severe heart failure: a randomised controlled trial. Br Heart J 71:146–150
Knauf H, Mutschler E (1995) Diuretic effectiveness of hydrochlorothiazide and furosemide alone and in combination in chronic renal failure. J Cardiovasc Pharmacol 26:394–400
Fliser D, Schroter M, Neubeck M, Ritz E (1994) Coadministration of thiazides increases the efficacy of loop diuretics even in patients with advanced renal failure. Kidney Int 46:482–488
Sica DA (2003) Metolazone and its role in edema management. Congest Heart Fail 9:100–105
Metolazone [package insert]; Mylan Pharmaceuticals Inc; Morgantown W. October 2004
Kissling KT, Pickworth KK (2014) Comparison of the effects of combination diuretic therapy with oral hydrochlorothiazide or intravenous chlorothiazide in patients receiving intravenous furosemide therapy for the treatment of heart failure. Pharmacotherapy. 34:882–887
Ter Maaten JM, Damman K (2018) Chloride, what else? Eur J Heart Fail 20:1444–1446
Grodin JL, Testani JM, Pandey A, Sambandam K, Drazner MH, Fang JC, Tang WHW (2018) Perturbations in serum chloride homeostasis in heart failure with preserved ejection fraction: insights from TOPCAT. Eur J Heart Fail 20:1436–1443
Cuthbert JJ, Pellicori P, Rigby A, Pan D, Kazmi S, Shah P, Clark AL (2018) Low serum chloride in patients with chronic heart failure: clinical associations and prognostic significance. Eur J Heart Fail 20:1426–1435
Ter Maaten JM, Damman K, Hanberg JS, Givertz MM, Metra M, O’Connor CM, Teerlink JR, Ponikowski P, Cotter G, Davison B, Cleland JG, Bloomfield DM, Hillege HL, van Veldhuisen DJ, Voors AA and Testani JM(2016) Hypochloremia, diuretic resistance, and outcome in patients with acute heart failure. Circ Heart Fail 9(8):e003109. https://doi.org/10.1161/CIRCHEARTFAILURE.116.003109
Hanberg JS, Rao V, Ter Maaten JM, Laur O, Brisco MA, Perry Wilson F, Grodin JL, Assefa M, Samuel Broughton J, Planavsky NJ, Ahmad T, Bellumkonda L, Tang WH, Parikh CR and Testani JM (2016) Hypochloremia and diuretic resistance in heart failure: mechanistic insights. Circ Heart Fail 9(8):e003180. https://doi.org/10.1161/CIRCHEARTFAILURE.116.003180
Ernst ME, Moser M (2009) Use of diuretics in patients with hypertension. N Engl J Med 361:2153–2164
Gines P, Cardenas A, Arroyo V, Rodes J (2004) Management of cirrhosis and ascites. N Engl J Med 350:1646–1654
European Association for the Study of the Liver (2010) EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol 53:397–417
Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J (1999) The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 341:709–717
Bansal S, Lindenfeld J, Schrier RW (2009) Sodium retention in heart failure and cirrhosis: potential role of natriuretic doses of mineralocorticoid antagonist? Circ Heart Fail 2:370–376
Butler J, Hernandez AF, Anstrom KJ, Kalogeropoulos A, Redfield MM, Konstam MA, Tang WH, Felker GM, Shah MR, Braunwald E (2016) Rationale and design of the ATHENA-HF trial: Aldosterone Targeted Neurohormonal Combined with Natriuresis Therapy in Heart Failure. JACC Heart Fail 4:726–735
Butler J, Anstrom KJ, Felker GM, Givertz MM, Kalogeropoulos AP, Konstam MA, Mann DL, Margulies KB, McNulty SE, Mentz RJ, Redfield MM, Tang WHW, Whellan DJ, Shah M, Desvigne-Nickens P, Hernandez AF, Braunwald E, National Heart Lung and Blood Institute Heart Failure Clinical Research Network (2017) Efficacy and safety of spironolactone in acute heart failure: the ATHENA-HF randomized clinical trial. JAMA Cardiol 2:950–958
Yang J, Young MJ (2016) Mineralocorticoid receptor antagonists—pharmacodynamics and pharmacokinetic differences. Curr Opin Pharmacol 27:78–85
Gheorghiade M, Konstam MA, Burnett JC Jr, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, Zimmer C, Orlandi C (2007) Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST clinical status trials. JAMA 297:1332–1343
Konstam MA, Gheorghiade M, Burnett JC Jr, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, Zimmer C, Orlandi C (2007) Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST outcome trial. JAMA 297:1319–1331
Gheorghiade M, Gattis WA, Barbagelata A, Adams KF Jr, Elkayam U, Orlandi C, O’Connor CM (2003) Rationale and study design for a multicenter, randomized, double-blind, placebo-controlled study of the effects of tolvaptan on the acute and chronic outcomes of patients hospitalized with worsening congestive heart failure. Am Heart J 145:S51–S54
Matsue Y, Suzuki M, Torii S, Yamaguchi S, Fukamizu S, Ono Y, Fujii H, Kitai T, Nishioka T, Sugi K, Onishi Y, Noda M, Kagiyama N, Satoh Y, Yoshida K, Goldsmith SR (2016) Clinical effectiveness of tolvaptan in patients with acute heart failure and renal dysfunction. J Card Fail 22:423–432
Felker GM, Mentz RJ, Cole RT, Adams KF, Egnaczyk GF, Fiuzat M, Patel CB, Echols M, Khouri MG, Tauras JM, Gupta D, Monds P, Roberts R, O’Connor CM (2017) Efficacy and safety of tolvaptan in patients hospitalized with acute heart failure. J Am Coll Cardiol 69:1399–1406
Konstam MA, Kiernan M, Chandler A, Dhingra R, Mody FV, Eisen H, Haught WH, Wagoner L, Gupta D, Patten R, Gordon P, Korr K, Fileccia R, Pressler SJ, Gregory D, Wedge P, Dowling D, Romeling M, Konstam JM, Massaro JM, Udelson JE, SECRET of CHF Investigators, Coordinators, and Committee Members (2017) Short-term effects of tolvaptan in patients with acute heart failure and volume overload. J Am Coll Cardiol 69:1409–1419
Mullens W, Verbrugge FH, Nijst P, Martens P, Tartaglia K, Theunissen E, Bruckers L, Droogne W, Troisfontaines P, Damman K, Lassus J, Mebazaa A, Filippatos G, Ruschitzka F, Dupont M (2018) Rationale and design of the ADVOR (Acetazolamide in Decompensated Heart Failure with Volume Overload) trial. Eur J Heart Fail 20:1591–1600
Knauf H, Mutschler E (1997) Sequential nephron blockade breaks resistance to diuretics in edematous states. J Cardiovasc Pharmacol 29:367–372
Verbrugge FH, Dupont M, Bertrand PB, Nijst P, Penders J, Dens J, Verhaert D, Vandervoort P, Tang WH, Mullens W (2015) Determinants and impact of the natriuretic response to diuretic therapy in heart failure with reduced ejection fraction and volume overload. Acta Cardiol 70:265–273
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cox, Z.L., Testani, J.M. Loop diuretic resistance complicating acute heart failure. Heart Fail Rev 25, 133–145 (2020). https://doi.org/10.1007/s10741-019-09851-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-019-09851-9