
Abstract
Despite significant progress in cardiovascular medicine, myocardial ischemia and infarction, progressing eventually to the final end point heart failure (HF), remain the leading cause of morbidity and mortality in the USA. HF is a complex syndrome that results from any structural or functional impairment in ventricular filling or blood ejection. Ultimately, the heart’s inability to supply the body’s tissues with enough blood may lead to death. Mechanistically, the hallmarks of the failing heart include abnormal energy metabolism, increased production of reactive oxygen species (ROS) and defects in excitation–contraction coupling. HF is a highly dynamic pathological process, and observed alterations in cardiac metabolism and function depend on the disease progression. In the early stages, cardiac remodeling characterized by normal or slightly increased fatty acid (FA) oxidation plays a compensatory, cardioprotective role. However, upon progression of HF, FA oxidation and mitochondrial oxidative activity are decreased, resulting in a significant drop in cardiac ATP levels. In HF, as a compensatory response to decreased oxidative metabolism, glucose uptake and glycolysis are upregulated, but this upregulation is not sufficient to compensate for a drop in ATP production. Elevated mitochondrial ROS generation and ROS-mediated damage, when they overwhelm the cellular antioxidant defense system, induce heart injury and contribute to the progression of HF. Mitochondrial uncoupling proteins (UCPs), which promote proton leak across the inner mitochondrial membrane, have emerged as essential regulators of mitochondrial membrane potential, respiratory activity and ROS generation. Although the physiological role of UCP2 and UCP3, expressed in the heart, has not been clearly established, increasing evidence suggests that these proteins by promoting mild uncoupling could reduce mitochondrial ROS generation and cardiomyocyte apoptosis and ameliorate thereby myocardial function. Further investigation on the alterations in cardiac UCP activity and regulation will advance our understanding of their physiological roles in the healthy and diseased heart and also may facilitate the development of novel and more efficient therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite significant progress in cardiovascular medicine, cardiovascular disease (CVD) remains the leading cause of combined morbidity and mortality in Western industrialized countries. According to the latest report from the American Heart Association, in the USA in 2008, CVD accounted for 32.8 % of all deaths. Major contributors are coronary artery disease, myocardial infarction (MI) and ischemic stroke, leading eventually to the final end point of heart failure (HF) [1, 2]. HF is a growing health problem, with a prevalence in the USA of almost 6 million, and is expected to reach 8.5 million by 2030 [3–6]. More than 1 million Americans with HF are hospitalized each year with a cost of approximately $40 billion/year [4]. Furthermore, the outcome for patients diagnosed with HF remains poor with approximately 50 % mortality within 4–5 years [7].
HF is ‘a complex syndrome that results from any structural or functional impairment in ventricular filling or ejection of blood’ [6]. Ultimately, this heart’s inability to supply the body’s tissues with enough blood may lead to death [8, 9]. Various types of heart damage, caused by myocardial ischemia, MI, pressure and work overload and genetic alterations, lead to cardiac remodeling progressing to HF [10–14]. The failing heart is characterized by impaired energy metabolism [15–20], increased production of reactive oxygen species (ROS) [21–24] and abnormal excitation–contraction coupling (ECC) [25–27].
HF is a highly dynamic pathological process, and observed alterations in substrate preference and energy metabolism depend on the disease progression [19, 20, 28–30]. In the early stages, cardiac remodeling plays an important compensatory, cardioprotective role with normal or slightly increased fatty acid (FA) β-oxidation (FAO), which provides 60–90 % of cardiac ATP production. However, upon progression of HF, FAO and mitochondrial respiratory activity decrease, resulting in cardiac ATP content reduction to 60–70 % of its physiological levels. As a compensatory response to decreased oxidative metabolism, glucose uptake and glycolysis are upregulated; however, this upregulation is not sufficient to compensate for the drop in ATP production [31–33].
Mitochondrial function is particularly important in the constantly energy demanding cardiomyocytes, in which mitochondria generate up to 90 % of cellular adenosine triphosphate (ATP) and occupy almost 1/3 of the cell volume of a cardiomyocyte [30, 34–37]. Over the past two decades, mitochondria have emerged not only as a powerhouse of the cell, but also as critical integrators of other essential cellular processes, such as cell death, contributing to health and disease [38–40]. Recent studies of cardiac mitochondria have convincingly demonstrated that the structural and functional alterations of these multifaceted organelles are implicated in the pathogenesis of various CVD, such as dysrhythmias, myocardial ischemia, cardiomyopathies and HF [15, 17, 41–47].
The double-membrane mitochondria use up to 90 % of O2 consumed by the cell to mediate oxidative phosphorylation (OXPHOS), the process coupling the substrate oxidation to ATP synthesis. In this process, the electrons released upon oxidation of NADH (nicotinamide adenine dinucleotide, reduced) and FADH2 (flavin adenine dinucleotide, reduced), the major products of the Krebs cycle, are transferred along the ‘respiratory chain,’ also known as the ‘electron transport chain’ (ETC), to O2, the terminal electron acceptor [48–50]. The ETC consists of four multi-subunit enzyme complexes I–IV, embedded in the inner mitochondrial membrane (IMM) and especially enriched in the cristae, and two soluble electron carriers, cytochrome c and coenzyme Q [51, 52]. According to Peter Mitchell’s chemiosmotic theory, proposed more than 50 years ago, the electron transfer generates a proton gradient (ΔpH) across the IMM (protons outside and hydroxyl ions inside). ΔpH along with the electrical gradient (ΔΨ m) forms the proton-motive force (Δp also denoted as Δμ H), which drives ATP synthesis from adenosine diphosphate (ADP) and inorganic phosphate (Pi) by the F1F0 ATP synthase (complex V) [53, 54].
However, OXPHOS is incompletely coupled, and protons can leak across the IMM and return to the mitochondrial matrix bypassing the F1F0 ATP synthase-mediated ATP production [54]. This proton leak can reach 20–70 % of the cellular metabolic rate in various cell types and depends on the presence of mitochondrial carrier proteins, the adenine nucleotide translocase (ANT) and uncoupling protein 1 (UCP1) in brown adipose tissue (BAT) [55–58].
Originally, UCP1-mediated proton conductance was believed to be a unique mechanism in BAT to generate heating, which Mitchell called ‘protic heating,’ evolutionarily acquired by mammals [54, 59–61]. However, it is currently well recognized that such proton leak uncoupling of OXPHOS occurs in other tissues, including the myocardium, and distinct UCP1 paralogues are present in various tissues and in all eukaryotic kingdoms: protists, fungi, plants and animals [62–65]. Growing evidence suggests that uncoupling proteins (UCPs) contribute to the regulation of mitochondrial ROS production associated with various disorders, including obesity, type 2 diabetes, insulin resistance, tumorigenesis, atherosclerosis, HF and aging [20, 64–67].
This review focuses on mitochondrial ROS generation in the healthy and failing heart and on the emerging role of UCPs in cardiac physiology and pathophysiology.
Oxidative stress and mitochondrial ROS production
In the heart, as one of the highest O2-consuming organs, highly tuned balance between O2 supply and consumption is vitally important to respond to physiological changes in workload and to pathological stresses, such as hypoxia, ischemia and excessive overload. ROS, including free radicals (e.g., superoxide [O .−2 ] and hydroxyl [.OH]), and non-radical species (e.g., hydrogen peroxide [H2O2]), and reactive nitrogen species (RNS) (e.g., nitric oxide [NO] and peroxynitrite [ONOO−]), are permanently generated from various intracellular sources [68–71].
Upon reduction of O2, one electron is added resulting in the formation of O .−2 ; the addition of the second electron converts O .−2 into non-radical H2O2. The reduction of H2O2 in the presence of endogenous Fe yields the generation of OH via the Fenton reaction. OH can also be generated through electron exchange between O .−2 and H2O2 in the Haber–Weiss reaction. O .−2 can also react with NO, leading to the production of ONOO−, the highly toxic lipid-soluble RNS capable to damage multiple molecules, leading eventually to cell dysfunction and death [70, 72–74]. At the molecular level, ROS/RNS mediate cysteine and methionine thiol oxidation, arginine and proline hydroxylation and tyrosine nitration of various proteins and modifications of other molecules affecting a variety of redox-sensitive process.
Under physiological conditions, ROS/RNS exert a critical role of second messengers, inducing multiple signaling pathways essential for cardiac function [23, 70, 75]. In cardiomyocytes, physiological levels of ROS/RNS can activate mitogen-activated protein kinases (MAPKs), such as the extracellular signal-regulated kinase 1/2 (ERK1/2), p38 and Jun N-terminal kinase (JNK) [76, 77], as well as other protective kinases, such as phosphatidylinositol 3-kinase (PI3K), protein kinase B/Akt and PKC [78], contributing to cardioprotection against ischemia/reperfusion (I/R) injury (IRI) [75, 79, 80].
However, when ROS/RNS generation is sharply increased and overwhelms the cellular antioxidant defense system, the condition known as oxidative stress (OS), they cause oxidative damage to a plethora of cellular macromolecules [23, 70, 75]. Among them are subunits of the ETC complexes I, III and V [81, 82], multiple myocardial proteins implicated in ECC, such as the ryanodine receptor 2 (RYR2) [83–86], myosin heavy chains [87], sarcoplasmic reticulum (SR) Ca2+-ATPase 2a (SERCA2a) [88–90], Ca2+/calmodulin-dependent kinase II (CaMKII) [91], cAMP-dependent protein kinase A (PKA) [92–94], cGMP-dependent protein kinase G 1α (PKG1α) [94, 95], and ion channels and transporters, such as L-type Ca channels, the plasmalemmal Ca2+-ATPase, the Na+/Ca2+ exchanger [96, 97]. S-nitrosylation of GAPDH and caspase-3 contributes to the initiation of hyperglycemia and cardiomyocyte death in the diabetic heart [98]. Oxidation of histone deacetylases (HDACs) and transcription factors nuclear factor-kB (NF-kB) and hypoxia-inducible factor 1 (HIF1) modulate the transcription in response to OS [99–102]. A recent list of ROS/RNS-modified key myocardial proteins found in diabetic cardiomyopathy includes 30 entries [103].
In addition to myocardial proteins, membrane lipids, cardiolipin (CL) in particular, are also subject to ROS-induced damage [104]. CL is a phospholipid predominantly localized in the IMM and implicated in assembly and function of the ETC, apoptotic signaling and mitochondrial protein import [105–107]. Oxidation of CL results in its pathogenic remodeling, affecting mitochondrial respiratory activity and triggering mitochondria-dependent apoptosis [106, 108–112]. Finally, excessive oxidative damage to mitochondrial DNA (mtDNA), if overwhelms the repair capacity, can cause severe mitochondrial dysfunction and eventually cardiomyocyte death [113–117]. Accumulation of oxidized and nitrated/nitrosylated proteins and lipids and oxidative lesions in mtDNA can lead to myocardial remodeling and dysfunction culminating eventually in HF (Fig. 1).

Oxidative stress in the myocardium. Excessive generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which overwhelms the capacity of antioxidant system, results in oxidative damage to proteins, DNA and lipids. Oxidative damaged cellular molecules affect a variety of essential cardiac processes, including energy metabolism, excitation–contracting coupling, Ca2+ homeostasis and cardiomyocyte death. These detrimental alterations lead to myocardial remodeling and dysfunction eventually culminating in heart failure (HF)
In cardiomyocytes, the main sources of endogenously generated ROS are mitochondria, NADPH oxidases (NOXs), uncoupled NO synthases (NOSs) and xanthine oxidase (XO). This review focuses on mitochondria-generated ROS/RNS and their role in the pathogenesis of HF. Role of NOXs, NOSs and XO in myocardial physiology and pathophysiology has been discussed in detail in recent excellent reviews [23, 70, 75, 118].
Mitochondrial ROS
In cardiomyocytes, mitochondria are the major source of ROS, which are generated as by-product of electron flow through the ETC, predominantly at complexes I and III [64, 69, 119]. It is believed that complex I produces O .−2 on the matrix side of the IMM, whereas complex III generates O .−2 on both the matrix and intermembrane sides of the IMM (Fig. 2). Complex I (NADH:ubiquinone oxidoreductase), composed of approximately 45 subunits, promotes oxidation of NADH to NAD. The electrons from NADH are accepted by the flavin mononucleotide (FMN) of complex I and are then transferred through a series of Fe–S clusters to ubiquinone (Q), resulting in its reduction to ubiquinol (QH2) [120]. The FMN prosthetic group in the soluble arm and the Q-binding site is mainly responsible for O .−2 generation by complex I [121–124].

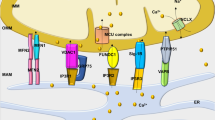
Cardiac mitochondria are the main source of reactive oxygen species (ROS). Superoxide (O .−2 ) is generated as by-product of electron flow through the electron transport chain (ETC), predominantly at complexes I and III. O .−2 can further be converted spontaneously or by action of mitochondrial Mn2+-dependent superoxide dismutase (MnSOD) to hydrogen peroxide (H2O2). NADPH oxidase 4 (NOX4), expressed in cardiomyocyte mitochondria, endoplasmic reticulum and nucleus, appears to generate predominantly H2O2 contributing to mitochondrial ROS. Monoamine oxidase (MAO), localized on the outer mitochondrial membrane (OMM), can also contribute to H2O2 production. The reduction of H2O2 in the presence of endogenous Fe2+ or Cu2+ yields the generation of hydroxyl radicals (.OH) and hydroxyl anions (OH−). Mitochondrial nitric oxide synthase (mtNOS), embedded in the inner mitochondrial membrane (IMM), can generate nitric oxide (NO), which rapidly reacts with O .−2 resulting in the production of peroxynitrite (ONOO−). Mitochondrial uncoupling proteins (UCP), located in the IMM, promote proton (H+) leak across the IMM from the intermembrane space (IMS) to the matrix, dissipating the proton gradient (ΔpH) and mitochondrial membrane potential (ΔΨ m ), generated by electron flow through the ETC, preventing excessive O .−2 generation
Complex III (ubiquinol:cytochrome c oxidoreductase), composed of 11 subunits, passes electrons from the ubiquinol produced by complex I and II to cytochrome c [125, 126]. The Q0 site of complex III is the major site responsible for ROS production [127]. Although the electron cycling process referred to the Q-cycle, in which lone electrons are reused to produce QH2, prevents to utilize these electrons for O .−2 production, complex III still remains the main source of ROS generation [127–130].
O .−2 produced by complex I and III can further be converted spontaneously or by action of mitochondrial Mn2+-dependent superoxide dismutase (MnSOD) or cytosolic Cu/ZnSOD to H2O2 [69, 71]. H2O2 is more stable than O .−2 and can cross membranes and oxidize glutathione (GSH) and thiol residues on various proteins, including kinases, phosphatases and other enzymes, as well as transcription factors [131]. Thus, H2O2 can modulate multiple signaling pathways essential for cell adaptation. However, excessively produced H2O2 results in the generation of highly reactive OH, which causes cell damage contributing to IRI [21].
It has recently been shown that NOX4, which is expressed in cardiomyocytes and generates predominantly H2O2, is located in the endoplasmic reticulum, nucleus and mitochondria and therefore can be consider as a source of mitochondrial ROS [75, 132]. Myocardial ischemia and chronic pressure overload have activated NOX4 expression in mouse hearts [133–135]. Transgenic mice with cardiac-specific NOX4 overexpression have exhibited compromised left-ventricular function and elevated apoptosis and fibrosis upon aging [136]. Consistently, cardiac-specific Nox4−/− mice have shown the reduced ROS levels in the heart and attenuated apoptosis associated with improved mitochondrial and cardiac function upon pressure overload compared with wild-type animals [137]. Importantly, preclinical studies of inhibitors specific for NOX4 and other NOX isoforms for treatment various cardiovascular conditions are in progress [138, 139].
In addition to the ETC-generated ROS on the IMM, monoamine oxidases (MAOs), which appear to be localized on the outer mitochondrial membrane (OMM), represent another potential mitochondrial source of ROS (Fig. 2) [140]. These enzymes, present in two isoforms, MAO-A and MAO-B, produce H2O2 and catalyze oxidative deamination of catecholamines and biogenic amines (e.g., epinephrine, norepinephrine and serotonin) [141]. Although the role of MAOs in the pathogenesis of human HF remains yet to be determined, it has recently been reported that MAO-A appears to contribute to adverse cardiac remodeling in a mouse pressure-overload HF model [142]. Functional role of MAOs in mitochondrial ROS production and the pathogenesis of HF has to be addressed in the future studies.
The presence of NOS in mitochondria was first reported by Bates et al. [143] and subsequently confirmed by other laboratories [144–147]. Despite some initial controversy regarding the identity of mitochondrial NOS (mtNOS) [148–152], current evidence suggests that mtNOS is a splicing variant of nNOS, embedded into the IMM (Fig. 2) [153–156]. Furthermore, the dependence of mtNOS activity on the function of the ETC complex I suggests their association [157, 158]. Consistent with this hypothesis, it has more recently been shown that inhibition of either complex I or II or mtNOS has led to reduction in ROS production in HF cardiomyocytes [159].
mtNOS generates NO that reacts rapidly with O .−2 , resulting in the production of ONOO−, a highly reactive short-lived peroxide, which can modify and inactivate several key mitochondrial proteins [160, 161]. ONOO− nitrates and inhibits activity of mitochondrial aconitase, an enzyme of Krebs cycle [162–164], and complexes I, II and V (ATP synthase), compromising mitochondrial bioenergetics [165, 166]. ONOO−-mediated nitration of tyrosine 34 (Tyr-34) on MnSOD inactivates this essential antioxidant amplifying mitochondrial OS [167–169]. MnSOD Tyr-34 nitration has been detected in various CVD [170, 171]. Nitration of Tyr-74 on cytochrome c has resulted in its translocation to the cytosol and nucleus and might be related to apoptotic response [172, 173]. Importantly, nitrated subunits of complex I and V and oxidized/nitrated MnSOD have been detected in diabetic failing hearts [174–176].
Antioxidant systems
In the heart, non-enzymatic and enzymatic antioxidative defense systems are involved in the control of ROS/RNS levels [70]. Non-enzymatic antioxidants include β-carotene (a precursor of vitamin A), vitamins C (ascorbic acid) and E (α-tocopherol), GSH, lipoic acid, ubiquinol (coenzyme Q-10), urate, polyamines and polyphenols and other substances [70, 177, 178]. Ascorbic acid and GSH are the main aqueous non-enzymatic scavengers playing a key role in cellular redox homeostasis. Intracellular GSH provides efficient protection against ONOO− and O .−2 , and cellular susceptibility to ONOO− largely depends on GSH abundance [73, 177].
Cytosolic and mitochondrial SODs, catalase (CAT) and the mitochondrial thioredoxin (Trx)/peroxiredoxin (Prx)/thioredoxin reductase and GSH/glutathione peroxidase (GPx) systems represent the best characterized antioxidant enzymes protecting the myocardium against OS [70, 179].
Superoxide dismutases (SODs)
SODs are key antioxidant enzymes, which catalyze the very fast conversion (2 × 109 M−1 s−1) of O .−2 into molecular O2 and H2O2 [180–182]. In humans, three types of SODs with regard to the metal cofactor they contain are known: Cu/ZnSOD, MnSOD and FeSOD. They have distinct structure and intracellular localization: Cu/ZnSOD is a homodimeric enzyme located in the cytosol (also known as SOD1 in humans) and in the extracellular space (also known as extracellular SOD or SOD3), while MnSOD (also known as SOD2) and FeSOD are homotetrameric enzymes located in the mitochondrial matrix and peroxisomes and in the extracellular space, respectively [183]. Mitochondrial MnSOD accounts for up to 90 % of total SOD activity in cardiomyocytes [70, 184].
Cu/ZnSOD deficiency in Sod1 −/− mice has resulted in high levels of oxidative damage associated with a significant decrease in lifespan compared with wild-type animals [185, 186]. Importantly, mutations in human gene encoded Cu/ZnSOD have been shown to be associated with neurodegenerative disorder amyotrophic lateral sclerosis (also known as Lou Gehrig’s disease) [187–189].
The essential role of mitochondrial MnSOD has been highlighted by studies on transgenic mice. Homozygous deletion of the Sod2 gene encoded MnSOD in mice has led to death within the first week of life with cardiomyopathy, degeneration of neurons, lipid accumulation in the liver and oxidative mitochondrial damage [190–193]. The most severely affected tissues have been the high-energy demanding heart and brain. Heterozygous Sod2 +/− mice have exhibited reduced MnSOD activity, elevated oxidative mtDNA damage in the heart and have developed cardiomyopathy during aging [114, 194]. Consistently, MnSOD overexpression has mediated cardioprotective effect against OS-induced cell death [195]. Furthermore, MnSOD deficiency has been shown to be associated with mtDNA damage and accelerate the development of atherosclerosis in ApoE−/− mice [196]. However, in contrast to data obtained from transgenic mice, current data on dynamics of cardiac SOD activity in patients with HF are controversial. Sam et al. [197] have reported decreased SOD activity associated with elevated ROS generation in the human failing heart, whereas others have failed to detect any significant changes in myocardial SOD levels or activities in patients with HF [198–200].
Catalase (CAT)
Mammalian CAT is a homotetrameric enzyme, which promotes the reduction of H2O2 to H2O and O2 [201–203]. As mammalian CAT is located in the peroxisomes and utilizes H2O2 generated during FAO in these organelles, it is thought to be not directly implicated in mitochondrial function. However, in some reports CAT has been found in cardiac mitochondria [204, 205]. Treatment of cells with H2O2 activates the Abelson (Abl) family of non-receptor tyrosine kinases, c-Abl and Arg, that phosphorylate CAT leading to its activation [206, 207]. To explore a role for CAT in OS response transgenic mice, which overexpress human CAT targeted to mitochondria (mCAT), have been generated [208]. mCAT animals have displayed reduced ROS production, oxidative mtDNA damage and deletion accumulation and extended life spans. Importantly, cardiac age-related alterations, including accumulation of mitochondrial protein oxidation, decreased cardiac SERCA2, increased mtDNA mutations and deletions and mitochondrial biogenesis, increased ventricular fibrosis, and enlarged myocardial fiber size, have significantly been attenuated in mCAT mice [209–211]. Intriguingly, CAT-deficient mice display no marked abnormalities, leaving question on the precise role of CAT in OS response open to debate [212].
Peroxiredoxins (Prxs) and glutathione peroxidases (GPxs)
Both Prxs and GPxs play an important role in redox state regulation by catalyzing the reduction of H2O2 to H2O and limiting thereby H2O2-induced OS [179, 213–215]. Among six mammalian Prx isoforms Prx3 and Prx5 are located in mitochondria. Prxs function through H2O2 mediated oxidation of its active cysteine site with subsequent reduction of the active site by Trx enabling Prxs to act as a H2O2 sensor [214, 215].
Mammalian cells express eight GPx isoforms, among them GPx1 is the predominant isoform and it is expressed in the myocardium. GPxs are tetrameric enzymes containing seleno-cysteine in the active site, which catalyze the reduction of H2O2 to H2O via oxidation of GSH into its disulfide form (GSSH) [179]. Similar to CAT, GPx1 can be phosphorylated and activated by c-Abl and Arg [216]. Ablation of GPx1 in mice causes increased susceptibility to H2O2-mediated OS and to myocardial IRI [217, 218]. Furthermore, GPx1 deficiency accelerates atherosclerotic progression in ApoE−/− mice [219].
The role of Prxs and GPxs in ROS scavenging depends on their relative abundance within mitochondria and on levels of ROS. Highly abundant Prxs appear to be responsible for conversion of low (nanomolar) levels of H2O2 under physiological conditions, while similarly active but less abundant GPxs can compete with Prxs at elevated H2O2 concentrations upon OS [214].
Uncoupling proteins: structure, regulation and function
Mammalian cells have evolved multiple mechanisms to tightly regulate levels of mitochondrial ROS. In addition to ROS scavengers as the first line of defense, inducible mitochondrial H+ leak across the IMM controlled by UCPs has emerged as an essential modulator of mitochondrial function. UCPs located in the IMM promote proton transport from the intermembrane space to the mitochondrial matrix dissipating ΔpH. This UCP-regulated mild uncoupling plays an important physiological role to avoid oversupply of electrons into the ETC adjusting energy metabolism and preventing excessive mitochondrial ROS generation [63–66, 220–222].
The UCP family
UCPs constitute a subfamily of the mitochondrial solute carrier 25 (SLC25) protein family, which contains over 40 members that mediate transport of broad range of molecules [223]. These ubiquitous eukaryotic trans-membrane proteins with molecular masses of 31–34 kDa are metabolite transporters, sharing similar molecular structure. UCP molecule is composed of six hydrophobic membrane-spanning α-helices, arranged into three cassettes. The amino and carboxyl termini and two loops, which connect cassettes, are oriented toward the intermembrane space, while three long loops that connect α-helices within each cassette face the matrix (Fig. 3) [63, 65, 224–227]. UCP α-helices appear to be arranged to create a channel within the IMM, while the loops are implicated in control of access to the channel [228]. Furthermore, UCPs contain also a binding site for purine nucleotides, which inhibit their uncoupling activity. Three arginine residues, Arg82, Arg182 and Arg276, conserved in UCP paralogues appear to play the crucial role in purine nucleotide binding [229].

Schematic representation of molecular structure and suggested physiological role of cardiac uncoupling protein 2 and 3 (UCP2/3). a Human UCPs share similar molecular structure. Six hydrophobic α-helices (1α through 6α), which span the inner mitochondrial membrane (IMM), are arranged into three cassettes. The N- and C-termini and two loops, which connect cassettes, are oriented toward the intermembrane space (IMS), while three long loops that connect α-helices within each cassette face the matrix. UCP α-helices appear to be arranged to create a channel within the IMM mediating proton (H+) leak across the IMM, while the loops control access to the channel. b Transcription factors (e.g., peroxisome proliferator-activated receptor [PPAR], thyroid hormone [TH], myogenic differentiation antigen [MyoD]), free fatty acids (FFA) and reactive oxygen species (ROS) regulate UCP expression in the heart. Although the physiological role of cardiac UCPs is not clearly defined, growing evidence suggests that they are involved in the regulation of energy metabolism, ROS generation, Ca2+ handling and cardiomyocyte apoptosis contributing to cardiac physiology and pathophysiology. See text for details
The first UCP, UCP1 (SLC25A7), was identified in BAT more than 30 years ago and has since become the canonical most characterized UCP [60, 230, 231]. UCP1 dissipates ΔpH to produce heat required to maintain body temperature in mammals [58, 232, 233]. Subsequently, four paralogues of UCP1, UCP2 through UCP5, have been indentified in fungal, plant and animal kingdoms [63, 226, 227, 234]. Mammalian cells express all five UCP paralogues, which share sequence similarities, but have different tissue distribution. UCP1 is predominantly expressed in BAT and upon hyperglycemia is also expressed in white adipose tissue, skeletal muscle, retinal cells and pancreatic β cells [58, 59, 235, 236]. Human UCP2 (SLC25A48) and UCP3 (SLC25A9) have high sequence identity with UCP1: 59 and 57 %, respectively, and ~70 % identity with each other [234]. UCP2 is widely expressed in various tissues, including central nervous system, kidney, macrophages, pancreas, spleen and thymus [237–239]; its expression in the heart is the issue of debate [240, 241]. UCP3 is predominantly expressed in skeletal muscle and BAT; it is also present in the heart albeit at lower levels compared with skeletal muscle [242–244]. Despite high sequence similarity of UCP2 and UCP3 with UCP1, they are not implicated in adaptive thermogenesis and their physiological role is largely unknown [63, 245–247].
UCP4 (SLC25A27) and UCP5 (SLC25A14; also known as BMCP1) have less sequence identity with UCP1: ~30 and 33 %, respectively; these UCPs are expressed mainly in the brain [63, 234, 248–252]. It has been hypothesized that the UCP4 and UCP5 genes are more closely related to the ancestral gene, which possibly encoded a primitive ADP/ATP transporter. Highly homologous to each other UCP1, UCP2 and UCP3 have evolved later during evolution [253]. Similar to UCP2 and UCP3, the physiological functions of human neuronal UCP4 and UCP5 remain yet largely uncertain [254].
UCP activity and its regulation
All mitochondrial UCPs promote H+ conductance dissipating ΔpH and affecting thereby mitochondrial function, although the precise mechanism of their action is still not fully understood. UCP1, which is highly abundant in BAT and implicated in the regulation of non-shivering thermogenesis, represents the archetypical UCP and our knowledge of the mechanism of the UCP-mediated proton conductance has mainly derived from studies of this protein. UCP1 is tightly regulated at several levels, including acute regulation of its uncoupling activity, transcriptional control of the Ucp1 gene expression and control of its degradation [65, 67].
Small molecules, such as FAs, ROS and purine nucleoside di- and tri-phosphates, exert acute regulation of UCP1 activity. Although the precise mechanism remains to be determined, three models of FA activation of the UCP-mediated proton leak have been proposed. According to the first ‘cofactor’ model, UCP1 functions as a H+ translocator, while anionic FAs (FA−) act as cofactors by associating with UCP1 and forming a H+-conducting channel [255–257]. In the second model, called the FA-cycling or flip-flop model, UCP1 functions as FA− carrier exporting FA− from the mitochondrial matrix. In the intermembrane space, exported FA− are protonated and diffuse back into the mitochondrial matrix, where the accepted protons are released [258]. Although according to this model UCP1 does not directly translocate H+, this cycle leads to a net H+ uptake into the matrix. Finally, given that the effect of FAs and purine nucleotides on H+ leak can be described by a simple competitive kinetics, the competition model suggests that FAs are not directly implicated in the H+ translocation, but rather act as allosteric activators influencing UCP1 conformation [259, 260].
At the molecular level, UCP1 activity is acutely stimulated not only by free FAs, but also by ROS (O .−2 and lipid peroxidation products) and inhibited by purine nucleoside di- and tri-phosphates, such as ADP, ATP, GDP and GTP [65–67, 261]. The mechanism of the acute ROS-mediated activation of UCPs is unclear and is currently debated. It has been suggested that O .−2 promotes peroxidation of membrane phospholipids resulting in the formation of 4-hydroxy-2-nonenal (4-HNE), which acts as a proximal activator of UCPs [262–265]. However, this model is not widely accepted and controversial observations exist [67, 247, 266–268].
Similarly, the precise mechanisms underlying inhibition of uncoupling activity by purine nucleotides remain to be determined. High affinity of UCPs to purine nucleotides with binding constants in the micromolar range and the millimolar concentrations of purine nucleotides in the cell raise the question how FAs can overcome this inhibition in vivo [63, 65, 260, 262, 269, 270].
More recently, distinct regulatory mechanisms for UCP1 and UCP2 and 3 have been suggested [64, 246]. Elevated ROS levels stimulate proton leak mediated by UCP2 and UCP3 leading subsequently to attenuation of ROS generation and creating a negative feedback loop [271, 272]. UCP1 is mainly activated by elevated free FAs in response to sympathetic neuronal stimulation resulting in mitochondria-promoted ROS generation [272, 273]. Furthermore, in vitro studies have shown that the reversible glutathionylation of UCP2 and UCP3 contributes to the regulation of mitochondrial ROS production [271]. Under OS condition, high ROS levels result in the glutathionylation of UCP2 and UCP3 inhibiting their uncoupling activity, while lower physiological levels of ROS lead to deglutathionylation of these proteins activating proton leak and attenuating thereby ROS production. Two cysteine residues Cys25 and Cys259, located in the first transmembrane region and the last loop facing the matrix, respectively, are main sites for the regulation of UCP3 function by glutathionylation. Given high similarities in amino acid sequence between UCP2 and UCP3, it has been suggested that UCP2 is regulated in a similar fashion [271]. Consistently, ROS-induced proton leak has been observed in the primary cells from wild-type but not from UCP2−/− or UCP3−/− mice [271]. Although UCP1 and UCP2/3 share high homology in cysteine residues, UCP1 activity appears to be not regulated by glutathionylation and is not involved in the control of mitochondrial ROS generation [271]. However, a physiological significance of this novel regulatory mechanism remains to be determined.
UCP levels are also highly regulated at the transcriptional level. In response to cold acclimation or chronic overfeeding, BAT sympathetic neurons release catecholamines (e.g., noradrenaline), which engage β3-adrenoceptors. Induced β3-adrenergic signaling activates adenylyl cyclase to produce cyclic AMP (cAMP). Elevated cAMP activates in turn PKA. PKA-promoted phosphorylation and activation of triacylglycerol lipase results in stimulation of lipolysis leading to the elevation of FA levels [274, 275]. Furthermore, cAMP-mediated signaling cascades control expression of the Ucp1 gene. A cAMP-responsive enhancer region upstream of the Ucp1 gene has binding sites for transcription factors belonging to the nuclear receptor family, such as the peroxisome proliferator-activated receptor γ (PPARγ), retinoic acid receptor (RXR) and thyroid hormone receptor (TR) [276–278].
The Ucp2 gene expression is also induced by PPARs as well as the sterol regulatory element-binding protein-1c (SREBP-1c) and forkhead transcription factors [279–283]. Furthermore, Ucp2 expression is upregulated by FAO and ROS contributing to cellular defense against OS [284–286]. Similarly, Ucp3 translation is stimulated by free FAs mediated through PPARs and the myogenic regulatory factor MyoD, thyroid hormone, retinoic acid and tumor necrosis factor α [287–291]. Furthermore, starvation-induced Ucp3 expression appears to be conserved in vertebrates including mammals [292, 293]. Finally, SIRT1, a protein involved in metabolic stress resistance, suppresses expression of both Ucp2 and Ucp3 genes and activates insulin secretion [294, 295].
Ucp2 and Ucp3 expression is also controlled at the translational level. The 5′ untranslated region of Ucp2 mRNA contains an inhibitory upstream open reading frame (ORF). Glutamine, an amino acid involved in insulin secretion, overcomes inhibitory effect of the ORF and upregulates Ucp2 mRNA translation [239, 296, 297]. Although Ucp3 mRNA translation is currently less studied, the 5′ untranslated region of Ucp3 mRNA also contains pseudo-start codons that can affect its translation [67].
Intracellular levels of UCPs are also regulated by proteolytic degradation. In BAT, UCP1 is relatively slowly degraded with a half-life on the order of days. Furthermore, noradrenergic stimulation upregulates not only UCP1 synthesis but also significantly extends its half-life [298, 299]. Although the precise mechanisms of the UCP1 turnover are largely unknown, its half-life is similar to those of other mitochondrial proteins, mirrors whole mitochondrial turnover and is extended upon lysosomal inhibition [299, 300].
In contrast to UCP1 and other mitochondrial carriers, including ANT, UCP2 and UCP3 are characterized by rapid degradation with very short half-lives of approximately 1 and 1–4 h for UCP2 and UCP3, respectively [239, 286, 301, 302]. Experiments with isolated energized mitochondria have suggested that non-mitochondrial factors are needed for rapid proteolytic degradation of these UCPs. Indeed, the cytosolic proteosome promotes rapid degradation of UCP2 and UCP3, while UCP1 and ANT are not degraded by this machinery [302, 303]. In fact, this has been the first demonstration that the IMM proteins are degraded by the cytosolic ubiquitin-proteosome system. Similar to highly coordinated control of UCP1 synthesis and degradation, cellular levels of UCP2 and UCP3 may be also regulated in a concerted manner [63, 67]. However, the precise mechanisms of these regulatory processes remain to be determined.
Importantly, the components involved in the insulin secretion pathway in the pancreatic β-cells are also under control of the cytosolic ubiquitin-proteosome system [304–306]. Therefore, rapid turnover of UCP2 and UCP3 expressed in the pancreatic β-cells may serve for rapid response to changes in nutrients in coordinated fashion with other proteins implicated in the same pathway [63, 67, 239]. Moreover, upregulation of UCP2, which has been detected in type 2 diabetes mellitus, may be linked to the proteosome system dysfunction [63, 307]. Physiological role and regulation of UCP3 and UCP2, which is particularly highly expressed in the pancreatic β-cells in type 2 diabetes mellitus, have been discussed in the several recent reviews [308–311].
Taken together, emerging distinct mechanisms underlying the regulation of UCP1 compared with UCP2 and UCP3 highlight the divergent physiological roles, which these proteins play. Function of UCP1 in the regulation of adaptive thermogenesis is well documented, whereas complex roles of UCP2 and UCP3 in the control of mitochondrial ROS generation and ROS signaling have to be further explored.
UCP 2 and UCP3 in the healthy and failing heart
It has already been discussed that HF as the terminal point of various CVD is characterized by abnormal bioenergetics and profound mitochondrial dysfunction, associated with excessive ROS production. Therefore, it is not surprising that several studies have focused on UCPs as regulators of these processes in the HF setting as well as on their use as potential therapeutic targets in treatment of HF. However, number of reports on UCP in various CVD is relatively limited and different laboratories have reached controversial conclusions regarding the functional roles of UCPs in cardiac physiology and pathophysiology.
In contrast to UCP1 present predominantly in BAT, highly homologous UCP2, found in various mammalian tissues, and UCP3 are expressed in the mammalian myocardium [237, 238, 240, 242, 312–314]. Cardiac UCP2 expression appears to be species specific; in the mouse heart, higher levels of UCP2 have been observed compared with those present in the human heart [237, 240]. UCP3 is a major UCP isoform expressed in skeletal muscle and in the heart, although its levels in the latter are lower [315]. The physiological role of UCP2 and UCP3 in the heart is not yet clearly defined; however, emerging evidence suggests that they are implicated in the regulation of cardiac energetics, mitochondrial ROS production, Ca2+ handling and cardiomyocyte death [241, 316, 317].
Several observations link cardiac UCPs, UCP3 in particular, and cardiac energy metabolism. Fasting and caloric restriction have been shown to modulate cardiac UCP3 expression. Fasting has induced a significant (1.5- to 3-fold) increase in UCP3 transcript levels in rat hearts [318, 319]. Similarly, caloric restriction has been associated with alterations in transcriptional profiles including upregulation of UCP3 expression in mouse hearts [320].
Thyroid hormone influences cardiac metabolism and bioenergetics [321]. It has been reported that thyroid hormone induces UCP2 and UCP3 expression at both mRNA and protein levels [317, 322–324]. Intriguingly, in the heart, thyroid hormone has upregulated UCP3 and possibly UCP2, but this upregulation has not been associated with increased respiratory uncoupling and inhibition of ATP production [325, 326]. Thus, the role of cardiac UCPs in the thyroid hormone-mediated heart energy metabolism remains to be determined.
Analyses of alterations in UCP2 and UCP3 expression in various cellular and animal models as well as in patients with CVD have provided to a certain degree contradictory data (Table 1). Some studies have demonstrated that both UCP2 and UCP3 are downregulated in the rat and human failing heart. In a rat model of HF induced by pressure overload, levels of UCP2 and UCP3 transcripts have been significantly decreased compared with control rats [319]. Furthermore, transcript levels of two other regulators of FAO and mitochondrial metabolism, pyruvate dehydrogenase kinase 4 (PDK4), malonyl-CoA decarboxylase (MCD), have also been reduced [327]. Expression of all these proteins are regulated by PPARα, consistently PPARα has been downregulated in hypertrophic and failing hearts [319, 327, 328]. The authors have suggested that downregulation of PPARα axis plays an adaptive cardioprotective role to prevent severe cardiac contractile dysfunction [327].
Metabolic expression profile has been analyzed in human fetal, non-failing (donor) and failing hearts. In failing hearts, the expression of UCP2 and to some extent UCP3 along with glucose transporter GLUT1 and GLUT4, energy metabolism enzymes (e.g., carnitine palmitoyl transferase, citrate synthase, pyruvate dehydrogenase kinase) and myosin heavy chains have been downregulated to the levels observed in fetal hearts [343]. Intriguingly, in the subsequent study, significantly reduced mRNA levels of UCP3, but not UCP2, have been detected in patients with HF awaiting heart transplantation upon placement and subsequent removal of a left ventricular assist device (LVAD) [344]. Importantly, UCP3 expression has been normalized with mechanical unloading after the LVAD treatment. Unfortunately, UCP2 and UCP3 protein levels have not been analyzed; therefore, it is not clear whether observed alterations in mRNA levels are translated into changes in protein concentrations.
Using a rat HF model caused by aortic regurgitation (AR), more complex dynamics of UCP2 expression in the failing heart has been shown. Initial decrease in UCP2 mRNA levels has changed to a significant increase in UCP2 expression in the late stage of HF [335]. Furthermore, upregulation of proinflammatory cytokine TNF-α has been detected in the late, but not early, stage of the development of HF. Authors have hypothesized that elevated TNF-α might be responsible for the induction of cardiac UCP2 expression in the late, chronic phase of HF. At the chronic stage of HF, increase in the cardiac expression of UCP2 has been accompanied by significant reduction of the high-energy phosphate, creatine phosphate (CrP), implying a decrease in energy efficiency in failing hearts [335]. In the follow-up study, authors have reported that elevated cardiac UCP2 expression, observed in the chronic stage of AR-induced HF, could be suppressed by the angiotensin-converting enzyme (ACE) inhibitor perindopril. Importantly, perindopril has also normalized CrP levels [336]. Unfortunately, as in previous studies, dynamics of UCP2 protein levels has been not analyzed.
More recently, in rat HF induced with intraperitoneal injections of chemotherapeutic agent doxorubicin, significant reduction in both UCP2 and UCP3 protein levels have been demonstrated [337]. Consistently, mitochondria isolated from failing hearts are characterized by greater coupling between citric acid cycle flux and ATP production. However, the beneficial effects of UCP2 and UCP3 downregulation on mitochondrial bioenergetics have been counteracted by augmented ROS generation observed in this HF model [337]. The mechanism responsible for UCP2 and UCP3 downregulation is also unknown.
It is well known that myocardial ischemia and infarction leading eventually to HF are associated with significant augmentation of circulating free FA levels [29, 345]. In contrast to UCP2 and UCP3 downregulation in HF observed in cited above reports, several studies have shown that elevated plasma free FA levels in HF are associated with upregulation of UCP2 and UCP3 in the failing heart. In animal models and in patients with HF, increased plasma long-chain FAs have activated PPARα leading to increased cardiac UCP mRNA and protein concentrations and reduced glucose transporter GLUT4 [240, 346–348]. Consistent with the regulatory role of PPARα, increased FA levels have been associated with UCP3 upregulation in wild-type mice but not in PPARα-deficient animals [348]. In the failing heart, elevated UCP2 and UCP3 increase mitochondrial uncoupling to attenuate ATP synthesis, while reduced GLUT4 downregulates glucose uptake. In addition, FAs may also act as activators of UCP activity further increasing mitochondrial uncoupling [349]. Similarly, in animal models of diabetic cardiomyopathy, elevated plasma free FA levels have been associated with significant upregulation of FA transporters (e.g., FATP and CD36) and UCP3 [325, 348, 350–354]. Moreover, significant increase in UCP2 mRNA levels, associated with reduced ATP production, has been detected in the Dahl salt-sensitive rat HF model [331].
In a rat model for chronically infarcted heart, UCP3 alterations along with mitochondrial respiration and efficiency of the isolated working heart have been measured [338]. Increased UCP3 levels in the failing heart have positively correlated with FA concentrations in the plasma. UCP3 upregulation has been associated with greater mitochondrial respiratory uncoupling and low efficiency in the failing heart [338]. Although augmented UCP-mediated respiratory uncoupling appears to underlie mitochondrial and cardiac dysfunction, it is not clear whether these alterations play adaptive or detrimental role in the development of HF.
Myocardial ischemia and infarction leading to HF are characterized by IRI, which affect cardiac energy metabolism, induce ROS generation, Ca2+ overload, acidosis and cardiomyocyte death [355, 356]. Mild mitochondrial uncoupling through UCP-mediated proton leak can play a protective role against myocardial IRI. One of the first direct evidence of a role of UCP3 in cardioprotection against I/R has recently been reported [333]. Ex vivo induced IRI in UPC3 −/− mouse hearts has resulted in poorer recovery of LV contractile function compared with wild-type mouse hearts under I/R conditions. Interestingly, isolated UPC2 −/− and wild-type mouse hearts have displayed a similar recovery of LV function, suggesting that UCP2 function is less essential for protection against cardiac IRI [333]. Using in vivo occlusion of the left coronary artery, these authors have further demonstrated that UPC3 −/− mice have twofold larger infarct size and higher incidence of I/R dysrhythmias than wild-type animals. Moreover, I/R has induced more severe alterations in cardiac energetics associated with more prominent increase in ROS generation in UPC3 −/− hearts compared with wild-type hearts. Pretreatment of UPC3-deficient hearts with the uncoupling drug carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone has ameliorated recovery after IRI. Finally, ischemic preconditioning has been completely abolished in UPC3-deficient hearts further confirming an essential role of UCP3 in cardioprotection against IRI [333].
In a porcine model of chronic myocardial ischemia, UCP2 protein levels have been found to be significantly increased within ischemic region, while UCP3 protein levels have not been changed. Importantly, mitochondria isolated from ischemic myocardium have displayed stress-resistant state characterized by mild uncoupling and reduced ROS production [341]. More recently, these authors using transgenic mouse model have demonstrated that UCP2 upregulation depends on the PPARγ-PGC1α axis. Chronic stimulation of PPARγ with its agonist pioglitazone has resulted in twofold increase in nuclear-located PGC1α and UCP2 levels [332]. Furthermore, isolated cardiac mitochondria with PPARγ-mediated UCP2 upregulation have displayed mild IMM depolarization and reduced ROS generation. These beneficial effects have not been detected in UPC2-deficient mice, suggesting that observed cardioprotection against IRI depends on UCP2 [332].
Acute MI in dog has led to significant upregulation (~1.7- and 3-fold 6 and 24 h post-infarction, respectively) of levels of UCP3 protein, the main cardiac UCP in dog [357], in the non-ischemic wall of the right ventricle (RV) [342]. Mitochondria within the non-ischemic wall have been uncoupled and levels of ROS have significantly been elevated. Authors have hypothesized that an increase in ROS induced by acute MI is responsible for the adaptive UCP3 upregulation [342].
It is well established that the heart can be protected against IRI by ischemic conditioning—brief repetitive cycles of ischemia delivered before or after the ischemic event [358–362]. Emerging evidence suggests that myocardial UCPs may be implicated in this complex process. It has recently been shown that cardiac ischemic preconditioning triggers upregulation of UCP2 and UCP3 on both mRNA and protein levels [330]. Mitochondria from preconditioned hearts have displayed increased proton leak associated with decreased ROS production. Significantly, UCP upregulation has been associated with reduced infarct size in preconditioned rat hearts. Furthermore, UCP depletion by RNA interference (RNAi) in rat cardiac H9c2 cells has resulted in attenuation of preconditioning of these cells and augmentation of ROS generation [330]. Similar ischemic preconditioning-induced upregulation of UCP2 have been demonstrated in the brain, where elevated UCP2 levels protect against neuronal ischemic injury possibly through attenuation of ROS generation [363, 364].
More recently, levels of cardiac UCP2 have been measured following acute cardiac I/R in rats. Acute myocardial I/R has induced significant increase in UCP2 protein levels in the ischemic area of the left ventricle (LV) but not in the RV [339]. Interestingly, the angiotensin type 1 receptor blocker losartan and the ACE inhibitor ramiprilat could suppress UCP2 expression induced by myocardial I/R protecting the heart against IRI. In the follow-up study, these authors have extended their analyses to UCP3 in cardiac I/R setting. Protein levels of both UCP2 and UCP3 have been significantly increased as an early response to acute myocardial I/R in rats [340]. However, the mechanism underplaying this upregulation appears to be different for UCP2 and UCP3. First, UCP2 has been upregulated only in ischemic area of the LV, while UCP3 has been increased in both ischemic area of the LV and non-ischemic region of the RV [340]. Authors have hypothesized that local upregulation of both UCP2 and UCP3 in ischemic region may be caused mainly by elevated ROS. A more global cardiac upregulation of UCP3 might be due to a higher responsiveness of UCP3 expression to elevated circulating free FAs observed in rats with HF [338]. Second, I/R-induced UCP2 upregulation has been shown on protein level, but not on UCP2 mRNA level, while upregulation of both UCP3 mRNA and protein has been detected [340].
Myocardial ischemia leading eventually to HF causes cardiomyocyte death resulting in significant cardiomyocyte loss, which represents the main prognostic parameter in the disease progression [365, 366]. Cardioprotective roles of UCPs against IRI may be linked to their involvement in cardiomyocyte death. UCP1 overexpression in cultured heart-derived H9c2 cells has limited ROS generation after I/R and prevented ROS-induced cell death preserving mitochondrial structure and function [367]. Consistently, using adenovirus-mediated transfection of cultured neonatal rat cardiomyocytes, it has been demonstrated that the overexpression of human UCP2 protects these cells from OS induced by H2O2 [329]. UCP2 overexpression has prevented mitochondrial membrane potential loss, ROS generation and Ca2+ overload. In addition, elevated UCP2 levels have attenuated the appearance of mitochondria-mediated apoptosis markers, such as TUNEL positivity, phosphatidyl serine exposure, propidium iodide uptake, and caspase-3 cleavage [329].
On the other hand, the overexpression of UCP2 in primary adult rat cardiomyocytes has led to controversial results [331]. Although UCP2 overexpression in these cells has resulted in significant decrease in ATP production and acidosis as well as in upregulation of pro-apoptotic protein BNIP3, cell survival at baseline has not been affected. However, UCP2-overexpressing cells have displayed lower survival after I/R compared with control cardiomyocytes. Furthermore, authors have demonstrated using a rat HF model the significant upregulation of UCP2 and BNIP3 in failing hearts [331]. Authors have concluded that under used experimental conditions UCP2 plays a detrimental role in cardiomyocyte survival in HF. However, they have suggested that it might have a protective effect under different conditions and/or in other species.
More recently, role of UCP3 in cell death induced by myocardial ischemia leading to HF has been studied using UPC3 −/− mice [334]. UPC3 −/− mouse embryonic fibroblasts and cardiomyocytes have displayed mitochondrial dysfunction, increased ROS generation and apoptosis under hypoxia. Infarct size has been larger in UPC3 −/− mice and these animals have exhibited lower survival compared with wild-type animals. Treatment with the antioxidant agent α-tocopherol has decreased infarct size in UPC3 −/− hearts to values found in wild-type hearts. UPC3 −/− hearts have been characterized by elevated of oxidative damage markers (e.g., TUNEL positive nuclei, p53 and cleaved caspase-3 levels). Finally, mitochondrial structural and functional abnormalities and elevated ROS production in ischemic UPC3 −/− hearts have occurred despite a normal UCP2 upregulation at mRNA and protein level [334]. Thus, these findings suggest a cardioprotective role of UCP3 in the ischemic heart.
Cardiac mitochondria play a critical role in Ca2+ handling in cardiomyocytes, which is vital for myocardial ECC and for proper cardiac function [368–370]. Data on the involvement of UCP2 and UCP3 in mitochondrial Ca2+ uniport activity in non-cardiac cells have been controversial [371, 372]. Using adenovirus-mediated UCP2 delivery into neonatal rat cardiomyocytes, Turner et al. [373] have demonstrated that UCP2 overexpression has suppressed mitochondrial Ca2+ uptake exerting thereby detrimental effects on beat-to-beat Ca2+ handling and ECC. Authors suggest that UCP2 upregulation observed in HF may enhance dysrhythmogenic potential and exacerbate contractile dysfunction contributing to the progression of the disease [373].
Finally, UCP2 protein may play a protective role against atherosclerosis, although data are so far limited. It has been reported that transplantation of bone marrow from UCP2 −/− mice into irradiated low-density lipoprotein receptor deficient (LDLR−/−) mice has led to significantly increased atherosclerotic lesion size when animals have been fed an atherogenic diet [374]. In addition, UCP2 −/− transplanted mice have displayed elevated levels of OS markers and the plaques from these animals have shown higher apoptosis. Consistent with a protective role of UCP2 in atherogenesis, the 866G/A and of a 45nt-del/ins polymorphism in the 3′-untranslated region of the UCP2 gene have been identified to be associated with carotid atherosclerosis in female study participants [375].
Conclusions
Despite great progress in our understanding of the molecular mechanisms underlying HF, this devastating disease represents a true challenge. Hallmarks of the failing heart are abnormal energy metabolism, increased production of ROS and defects in ECC. HF is a highly dynamic pathological process, and observed alterations in cardiac metabolism and function depend on the disease progression.
Mitochondrial ROS generation and ROS-mediated damage clearly contribute to the development and progression of HF. Cardiac UCP2 and UCP3 promoting proton leak across the IMM have emerged as essential regulators of mitochondrial membrane potential and respiratory function. However, unlike the well established role of their homolog UCP1 in adaptive thermogenesis in BAT, the physiological function of UCP2 and UCP3 in the heart is not clearly understood. Growing evidence suggests that cardiac UCPs are able to promote mild uncoupling reducing excessive mitochondrial ROS generation and ameliorating thereby myocardial function.
Controversial data have been reported regarding alterations in the expression of UCP2 and UCP3 seen in different animal HF models. Poorly understood variability in animal models of HF combined with species-specific UCP expression pattern might be responsible at least in part for these controversies. Unfortunately, it is even less known the alterations in UCP2 and UCP3 expression in patients with HF. Nevertheless, more recent studies suggest an upregulation of UCP2 or UCP3, leading to mitochondrial uncoupling and reduced ROS generation especially in ischemia-induced HF (Table 1). Since UCP2 and/or UCP3 can limit excessive mitochondrial ROS generation reducing the efficiency of ATP synthesis, it is unclear under which conditions their function would be protective or deleterious in the development of HF.
Similarly, although emerging evidence suggests that cardiac UCPs contribute to triggering cardiomyocyte death and ECC dysfunction in the failing heart, the underlying mechanisms remain largely undefined. For example, upregulated cardiac UCPs appear to trigger the expression of several apoptotic markers; however, this does not necessarily affect cardiomyocyte survival. Finally, it remains possible that the observed alterations in cardiac UCPs levels and/or activity are secondary to pathological cardiac remodeling. Future research efforts should address these critical issues.
High-throughput screening of mutations and polymorphisms in the genes encoding these proteins associated with increased risk for CVD is a novel promising approach. Recent identification of several polymorphisms in the UCP1, UCP2 and UCP3 genes associated with diabetes mellitus represents a successful example of such approach [311]. As discussed above, this approach has successfully been applied to identifying polymorphisms in the UCP3 gene associated with atherosclerosis.
In summary, further research on UCPs activity and regulation will be necessary to advance our understanding of their function in the healthy and diseased heart. Moreover, the combined effort of molecular and clinical cardiologists is needed before we can use cardiac UCPs as targets to treat HF.
References
Nabel EG, Braunwald E (2012) A tale of coronary artery disease and myocardial infarction. N Engl J Med 366:54–63
Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD et al (2012) Executive summary: heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 125:188–197
Braunwald E (2013) Research advances in heart failure: a compendium. Circ Res 113:633–645
Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J et al (2013) Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail 6:606–619
Roger VL (2013) Epidemiology of heart failure. Circ Res 113:646–659
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr et al (2013) 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 62:e147–e239
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD et al (2013) Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 127:e6–e245
Mudd JO, Kass DA (2008) Tackling heart failure in the twenty-first century. Nature 451:919–928
Shah AM, Mann DL (2011) In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet 378:704–712
McMurray JJ, Pfeffer MA (2005) Heart failure. Lancet 365:1877–1889
Monnet E, Chachques JC (2005) Animal models of heart failure: What is new? Ann Thorac Surg 79:1445–1453
Klocke R, Tian W, Kuhlmann MT, Nikol S (2007) Surgical animal models of heart failure related to coronary heart disease. Cardiovasc Res 74:29–38
Zornoff LA, Paiva SA, Duarte DR, Spadaro J (2009) Ventricular remodeling after myocardial infarction: concepts and clinical implications. Arq Bras Cardiol 92:150–164
McMurray JJ (2010) Clinical practice. Systolic heart failure. N Engl J Med 362:228–238
Neubauer S (2007) The failing heart—an engine out of fuel. N Engl J Med 356:1140–1151
Ingwall JS (2009) Energy metabolism in heart failure and remodelling. Cardiovasc Res 81:412–419
Rosca MG, Hoppel CL (2010) Mitochondria in heart failure. Cardiovasc Res 88:40–50
Abel ED, Doenst T (2011) Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 90:234–242
Azevedo PS, Minicucci MF, Santos PP, Paiva SA, Zornoff LA (2013) Energy metabolism in cardiac remodeling and heart failure. Cardiol Rev 21:135–140
Nickel A, Loffler J, Maack C (2013) Myocardial energetics in heart failure. Basic Res Cardiol 108:358
Giordano FJ (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115:500–508
Maack C, Bohm M (2011) Targeting mitochondrial oxidative stress in heart failure throttling the afterburner. J Am Coll Cardiol 58:83–86
Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM (2011) Redox signaling in cardiac myocytes. Free Radic Biol Med 50:777–793
Chen AF, Chen DD, Daiber A, Faraci FM, Li H et al (2012) Free radical biology of the cardiovascular system. Clin Sci (Lond) 123:73–91
Houser SR, Margulies KB (2003) Is depressed myocyte contractility centrally involved in heart failure? Circ Res 92:350–358
Bers DM (2006) Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 21:380–387
Neef S, Maier LS (2013) Novel aspects of excitation–contraction coupling in heart failure. Basic Res Cardiol 108:360
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90:207–258
Ventura-Clapier R, Garnier A, Veksler V, Joubert F (2011) Bioenergetics of the failing heart. Biochim Biophys Acta 1813:1360–1372
Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP et al (2005) Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation 112:2339–2346
Neglia D, De Caterina A, Marraccini P, Natali A, Ciardetti M et al (2007) Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 293:H3270–H3278
Kolwicz SC Jr, Tian R (2011) Glucose metabolism and cardiac hypertrophy. Cardiovasc Res 90:194–201
Ingwall JS (2002) ATP and the heart. Kluwer Academic, Norwell, MA
Hoppel CL, Tandler B, Fujioka H, Riva A (2009) Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol 41:1949–1956
Lemieux H, Hoppel CL (2009) Mitochondria in the human heart. J Bioenerg Biomembr 41:99–106
Ong SB, Hausenloy DJ (2010) Mitochondrial morphology and cardiovascular disease. Cardiovasc Res 88:16–29
Soubannier V, McBride HM (2009) Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta 1793:154–170
Hausenloy DJ, Ruiz-Meana M (2010) Not just the powerhouse of the cell: emerging roles for mitochondria in the heart. Cardiovasc Res 88:5–6
Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148:1145–1159
Brown DA, O’Rourke B (2010) Cardiac mitochondria and arrhythmias. Cardiovasc Res 88:241–249
Cadenas S, Aragones J, Landazuri MO (2010) Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc Res 88:219–228
Rosca MG, Hoppel CL (2009) New aspects of impaired mitochondrial function in heart failure. J Bioenerg Biomembr 41:107–112
Balaban RS (2012) Perspectives on: SGP symposium on mitochondrial physiology and medicine: metabolic homeostasis of the heart. J Gen Physiol 139:407–414
Verdejo HE, del Campo A, Troncoso R, Gutierrez T, Toro B et al (2012) Mitochondria, myocardial remodeling, and cardiovascular disease. Curr Hypertens Rep 14:532–539
Dorn GW 2nd (2013) Mitochondrial dynamics in heart disease. Biochim Biophys Acta 1833:233–241
Ong SB, Hall AR, Hausenloy DJ (2013) Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Signal 19:400–414
Saraste M (1999) Oxidative phosphorylation at the fin de siecle. Science 283:1488–1493
Balaban RS (2009) Domestication of the cardiac mitochondrion for energy conversion. J Mol Cell Cardiol 46:832–841
Brand MD, Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J 435:297–312
Genova ML, Bianchi C, Lenaz G (2003) Structural organization of the mitochondrial respiratory chain. Ital J Biochem 52:58–61
Benard G, Rossignol R (2008) Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid Redox Signal 10:1313–1342
Mitchell P (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191:144–148
Mitchell P (1979) Keilin’s respiratory chain concept and its chemiosmotic consequences. Science 206:1148–1159
Rolfe DF, Brand MD (1997) The physiological significance of mitochondrial proton leak in animal cells and tissues. Biosci Rep 17:9–16
Affourtit C, Brand MD (2006) Stronger control of ATP/ADP by proton leak in pancreatic beta-cells than skeletal muscle mitochondria. Biochem J 393:151–159
Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC et al (2005) The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J 392:353–362
Parker N, Crichton PG, Vidal-Puig AJ, Brand MD (2009) Uncoupling protein-1 (UCP1) contributes to the basal proton conductance of brown adipose tissue mitochondria. J Bioenerg Biomembr 41:335–342
Klingenberg M (1990) Mechanism and evolution of the uncoupling protein of brown adipose tissue. Trends Biochem Sci 15:108–112
Nicholls DG, Rial E (1999) A history of the first uncoupling protein, UCP1. J Bioenerg Biomembr 31:399–406
Klingenberg M, Echtay KS (2001) Uncoupling proteins: the issues from a biochemist point of view. Biochim Biophys Acta 1504:128–143
Sluse FE, Jarmuszkiewicz W (2002) Uncoupling proteins outside the animal and plant kingdoms: functional and evolutionary aspects. FEBS Lett 510:117–120
Azzu V, Brand MD (2010) The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci 35:298–307
Mailloux RJ, Harper ME (2011) Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med 51:1106–1115
Sluse FE (2012) Uncoupling proteins: molecular, functional, regulatory, physiological and pathological aspects. Adv Exp Med Biol 942:137–156
Echtay KS (2007) Mitochondrial uncoupling proteins–what is their physiological role? Free Radic Biol Med 43:1351–1371
Azzu V, Jastroch M, Divakaruni AS, Brand MD (2010) The regulation and turnover of mitochondrial uncoupling proteins. Biochim Biophys Acta 1797:785–791
Massion PB, Balligand JL (2007) Relevance of nitric oxide for myocardial remodeling. Curr Heart Fail Rep 4:18–25
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Nediani C, Raimondi L, Borchi E, Cerbai E (2011) Nitric oxide/reactive oxygen species generation and nitroso/redox imbalance in heart failure: from molecular mechanisms to therapeutic implications. Antioxid Redox Signal 14:289–331
Raedschelders K, Ansley DM, Chen DD (2012) The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol Ther 133:230–255
Brown GC, Borutaite V (2007) Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res 75:283–290
Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424
Radi R (2013) Peroxynitrite, a stealthy biological oxidant. J Biol Chem 288:26464–26472
Zhang Y, Tocchetti CG, Krieg T, Moens AL (2012) Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radic Biol Med 53:1531–1540
Sabri A, Byron KL, Samarel AM, Bell J, Lucchesi PA (1998) Hydrogen peroxide activates mitogen-activated protein kinases and Na+–H+ exchange in neonatal rat cardiac myocytes. Circ Res 82:1053–1062
Wei S, Rothstein EC, Fliegel L, Dell’Italia LJ, Lucchesi PA (2001) Differential MAP kinase activation and Na(+)/H(+) exchanger phosphorylation by H(2)O(2) in rat cardiac myocytes. Am J Physiol Cell Physiol 281:C1542–C1550
Sabri A, Hughie HH, Lucchesi PA (2003) Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid Redox Signal 5:731–740
Hausenloy DJ, Wynne AM, Yellon DM (2007) Ischemic preconditioning targets the reperfusion phase. Basic Res Cardiol 102:445–452
Dost T, Cohen MV, Downey JM (2008) Redox signaling triggers protection during the reperfusion rather than the ischemic phase of preconditioning. Basic Res Cardiol 103:378–384
Baseler WA, Dabkowski ER, Williamson CL, Croston TL, Thapa D et al (2011) Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: contribution of protein import dysfunction. Am J Physiol Regul Integr Comp Physiol 300:R186–R200
Hollander JM, Baseler WA, Dabkowski ER (2011) Proteomic remodeling of mitochondria in heart failure. Congest Heart Fail 17:262–268
Eager KR, Roden LD, Dulhunty AF (1997) Actions of sulfhydryl reagents on single ryanodine receptor Ca(2+)-release channels from sheep myocardium. Am J Physiol 272:C1908–C1918
Marengo JJ, Hidalgo C, Bull R (1998) Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys J 74:1263–1277
Zissimopoulos S, Lai FA (2006) Redox regulation of the ryanodine receptor/calcium release channel. Biochem Soc Trans 34:919–921
Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A et al (2008) Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res 103:1466–1472
Shao CH, Rozanski GJ, Nagai R, Stockdale FE, Patel KP et al (2010) Carbonylation of myosin heavy chains in rat heart during diabetes. Biochem Pharmacol 80:205–217
Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS et al (2004) S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 10:1200–1207
Sharov VS, Dremina ES, Galeva NA, Williams TD, Schoneich C (2006) Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J 394:605–615
Tang WH, Kravtsov GM, Sauert M, Tong XY, Hou XY et al (2010) Polyol pathway impairs the function of SERCA and RyR in ischemic-reperfused rat hearts by increasing oxidative modifications of these proteins. J Mol Cell Cardiol 49:58–69
Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J et al (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133:462–474
Humphries KM, Juliano C, Taylor SS (2002) Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem 277:43505–43511
de Pina MZ, Vazquez-Meza H, Pardo JP, Rendon JL, Villalobos-Molina R et al (2008) Signaling the signal, cyclic AMP-dependent protein kinase inhibition by insulin-formed H2O2 and reactivation by thioredoxin. J Biol Chem 283:12373–12386
Burgoyne JR, Eaton P (2009) Transnitrosylating nitric oxide species directly activate type I protein kinase A, providing a novel adenylate cyclase-independent cross-talk to beta-adrenergic-like signaling. J Biol Chem 284:29260–29268
Prysyazhna O, Rudyk O, Eaton P (2012) Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med 18:286–290
Zima AV, Blatter LA (2006) Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71:310–321
Hool LC, Corry B (2007) Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid Redox Signal 9:409–435
Puthanveetil P, Zhang D, Wang Y, Wang F, Wan A et al (2012) Diabetes triggers a PARP1 mediated death pathway in the heart through participation of FoxO1. J Mol Cell Cardiol 53:677–686
Ago T, Liu T, Zhai P, Chen W, Li H et al (2008) A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133:978–993
Loor G, Schumacker PT (2008) Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ 15:686–690
Rey S, Semenza GL (2010) Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res 86:236–242
Burgoyne JR, Mongue-Din H, Eaton P, Shah AM (2012) Redox signaling in cardiac physiology and pathology. Circ Res 111:1091–1106
Varga ZV, Giricz Z, Liaudet L, Hasko G, Ferdinandy P et al (2014) Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta. http://www.sciencedirect.com/science/article/pii/S0925443914002075
Yin H, Xu L, Porter NA (2011) Free radical lipid peroxidation: mechanisms and analysis. Chem Rev 111:5944–5972
Houtkooper RH, Vaz FM (2008) Cardiolipin, the heart of mitochondrial metabolism. Cell Mol Life Sci 65:2493–2506
Sparagna GC, Lesnefsky EJ (2009) Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol 53:290–301
Claypool SM, Koehler CM (2012) The complexity of cardiolipin in health and disease. Trends Biochem Sci 37:32–41
Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A et al (2004) Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 94:53–59
Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ (2008) Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 294:C460–C466
Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA et al (2007) Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res 48:1559–1570
Yin H, Zhu M (2012) Free radical oxidation of cardiolipin: chemical mechanisms, detection and implication in apoptosis, mitochondrial dysfunction and human diseases. Free Radic Res 46:959–974
Kagan VE, Chu CT, Tyurina YY, Cheikhi A, Bayir H (2014) Cardiolipin asymmetry, oxidation and signaling. Chem Phys Lipids 179:64–69
Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K et al (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481–484
Loch T, Vakhrusheva O, Piotrowska I, Ziolkowski W, Ebelt H et al (2009) Different extent of cardiac malfunction and resistance to oxidative stress in heterozygous and homozygous manganese-dependent superoxide dismutase-mutant mice. Cardiovasc Res 82:448–457
Pohjoismaki JL, Goffart S, Taylor RW, Turnbull DM, Suomalainen A et al (2010) Developmental and pathological changes in the human cardiac muscle mitochondrial DNA organization, replication and copy number. PLoS ONE 5:e10426
Pohjoismaki JL, Boettger T, Liu Z, Goffart S, Szibor M et al (2012) Oxidative stress during mitochondrial biogenesis compromises mtDNA integrity in growing hearts and induces a global DNA repair response. Nucl Acids Res 40:6595–6607
Copeland WC, Longley MJ (2014) Mitochondrial genome maintenance in health and disease. DNA Repair (Amst) 19:190–198
Kleikers PW, Wingler K, Hermans JJ, Diebold I, Altenhofer S et al (2012) NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med (Berl) 90:1391–1406
Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE (2009) Mitochondria and reactive oxygen species. Free Radic Biol Med 47:333–343
Hirst J, Carroll J, Fearnley IM, Shannon RJ, Walker JE (2003) The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim Biophys Acta 1604:135–150
Turrens JF, Boveris A (1980) Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 191:421–427
Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS (2004) Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem 279:4127–4135
Lambert AJ, Brand MD (2004) Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J Biol Chem 279:39414–39420
Treberg JR, Quinlan CL, Brand MD (2011) Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem 286:27103–27110
Miwa S, Brand MD (2005) The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim Biophys Acta 1709:214–219
Liu SS (2010) Mitochondrial Q cycle-derived superoxide and chemiosmotic bioenergetics. Ann N Y Acad Sci 1201:84–95
Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP et al (2007) The Q0 site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177:1029–1036
Crofts AR, Holland JT, Victoria D, Kolling DR, Dikanov SA et al (2008) The Q-cycle reviewed: How well does a monomeric mechanism of the bc(1) complex account for the function of a dimeric complex? Biochim Biophys Acta 1777:1001–1019
Turrens JF, Freeman BA, Levitt JG, Crapo JD (1982) The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch Biochem Biophys 217:401–410
Klimova T, Chandel NS (2008) Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ 15:660–666
Groeger G, Quiney C, Cotter TG (2009) Hydrogen peroxide as a cell-survival signaling molecule. Antioxid Redox Signal 11:2655–2671
Maejima Y, Kuroda J, Matsushima S, Ago T, Sadoshima J (2011) Regulation of myocardial growth and death by NADPH oxidase. J Mol Cell Cardiol 50:408–416
Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C et al (2003) Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 93:802–805
Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N et al (2008) Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 51:319–325
Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ et al (2010) NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA 107:18121–18126
Serrander L, Cartier L, Bedard K, Banfi B, Lardy B et al (2007) NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406:105–114
Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD et al (2010) NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA 107:15565–15570
Altenhofer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ et al (2012) The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci 69:2327–2343
Altenhofer S, Radermacher KA, Kleikers PW, Wingler K, Schmidt HH (2014) Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal. http://online.liebertpub.com/doi/pdf/10.1089/ars.2013.5814
Mialet-Perez J, Bianchi P, Kunduzova O, Parini A (2007) New insights on receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. J Neural Transm 114:823–827
Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F (2004) Structure and mechanism of monoamine oxidase. Curr Med Chem 11:1983–1993
Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW et al (2010) Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res 106:193–202
Bates TE, Loesch A, Burnstock G, Clark JB (1995) Immunocytochemical evidence for a mitochondrially located nitric oxide synthase in brain and liver. Biochem Biophys Res Commun 213:896–900
Ghafourifar P, Richter C (1997) Nitric oxide synthase activity in mitochondria. FEBS Lett 418:291–296
Giulivi C, Poderoso JJ, Boveris A (1998) Production of nitric oxide by mitochondria. J Biol Chem 273:11038–11043
Tatoyan A, Giulivi C (1998) Purification and characterization of a nitric-oxide synthase from rat liver mitochondria. J Biol Chem 273:11044–11048
Carreras MC, Peralta JG, Converso DP, Finocchietto PV, Rebagliati I et al (2001) Modulation of liver mitochondrial NOS is implicated in thyroid-dependent regulation of O(2) uptake. Am J Physiol Heart Circ Physiol 281:H2282–H2288
Bates TE, Loesch A, Burnstock G, Clark JB (1996) Mitochondrial nitric oxide synthase: a ubiquitous regulator of oxidative phosphorylation? Biochem Biophys Res Commun 218:40–44
Frandsen U, Lopez-Figueroa M, Hellsten Y (1996) Localization of nitric oxide synthase in human skeletal muscle. Biochem Biophys Res Commun 227:88–93
Koivisto A, Matthias A, Bronnikov G, Nedergaard J (1997) Kinetics of the inhibition of mitochondrial respiration by NO. FEBS Lett 417:75–80
Carreras MC, Melani M, Riobo N, Converso DP, Gatto EM et al (2002) Neuronal nitric oxide synthases in brain and extraneural tissues. Methods Enzymol 359:413–423
Valdez LB, Zaobornyj T, Alvarez S, Bustamante J, Costa LE et al (2004) Heart mitochondrial nitric oxide synthase. Effects of hypoxia and aging. Mol Aspects Med 25:49–59
Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM et al (2001) Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc Natl Acad Sci USA 98:14126–14131
Elfering SL, Sarkela TM, Giulivi C (2002) Biochemistry of mitochondrial nitric-oxide synthase. J Biol Chem 277:38079–38086
Finocchietto PV, Franco MC, Holod S, Gonzalez AS, Converso DP et al (2009) Mitochondrial nitric oxide synthase: a masterpiece of metabolic adaptation, cell growth, transformation, and death. Exp Biol Med (Maywood) 234:1020–1028
Lacza Z, Pankotai E, Busija DW (2009) Mitochondrial nitric oxide synthase: current concepts and controversies. Front Biosci 14:4436–4443
Parihar MS, Nazarewicz RR, Kincaid E, Bringold U, Ghafourifar P (2008) Association of mitochondrial nitric oxide synthase activity with respiratory chain complex I. Biochem Biophys Res Commun 366:23–28
Navarro A, Bandez MJ, Gomez C, Repetto MG, Boveris A (2010) Effects of rotenone and pyridaben on complex I electron transfer and on mitochondrial nitric oxide synthase functional activity. J Bioenerg Biomembr 42:405–412
Dedkova EN, Seidlmayer LK, Blatter LA (2013) Mitochondria-mediated cardioprotection by trimetazidine in rabbit heart failure. J Mol Cell Cardiol 59:41–54
Radi R, Cassina A, Hodara R, Quijano C, Castro L (2002) Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33:1451–1464
Castro L, Demicheli V, Tortora V, Radi R (2011) Mitochondrial protein tyrosine nitration. Free Radic Res 45:37–52
Castro L, Rodriguez M, Radi R (1994) Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J Biol Chem 269:29409–29415
Hausladen A, Fridovich I (1994) Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem 269:29405–29408
Tortora V, Quijano C, Freeman B, Radi R, Castro L (2007) Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radic Biol Med 42:1075–1088
Radi R, Rodriguez M, Castro L, Telleri R (1994) Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys 308:89–95
Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F et al (1996) Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328:85–92
MacMillan-Crow LA, Crow JP, Thompson JA (1998) Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 37:1613–1622
Yamakura F, Taka H, Fujimura T, Murayama K (1998) Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J Biol Chem 273:14085–14089
Moreno DM, Marti MA, De Biase PM, Estrin DA, Demicheli V et al (2011) Exploring the molecular basis of human manganese superoxide dismutase inactivation mediated by tyrosine 34 nitration. Arch Biochem Biophys 507:304–309
Xu S, Ying J, Jiang B, Guo W, Adachi T et al (2006) Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol 290:H2220–H2227
Redondo-Horcajo M, Romero N, Martinez-Acedo P, Martinez-Ruiz A, Quijano C et al (2010) Cyclosporine A-induced nitration of tyrosine 34 MnSOD in endothelial cells: role of mitochondrial superoxide. Cardiovasc Res 87:356–365
Abriata LA, Cassina A, Tortora V, Marin M, Souza JM et al (2009) Nitration of solvent-exposed tyrosine 74 on cytochrome c triggers heme iron-methionine 80 bond disruption. Nuclear magnetic resonance and optical spectroscopy studies. J Biol Chem 284:17–26
Godoy LC, Munoz-Pinedo C, Castro L, Cardaci S, Schonhoff CM et al (2009) Disruption of the M80-Fe ligation stimulates the translocation of cytochrome c to the cytoplasm and nucleus in nonapoptotic cells. Proc Natl Acad Sci USA 106:2653–2658
Turko IV, Li L, Aulak KS, Stuehr DJ, Chang JY et al (2003) Protein tyrosine nitration in the mitochondria from diabetic mouse heart. Implications to dysfunctional mitochondria in diabetes. J Biol Chem 278:33972–33977
Cong W, Zhao T, Zhu Z, Huang B, Ma W et al (2014) Metallothionein prevents cardiac pathological changes in diabetes by modulating nitration and inactivation of cardiac ATP synthase. J Nutr Biochem 25:463–474
Dennis KE, Hill S, Rose KL, Sampson UK, Hill MF (2013) Augmented cardiac formation of oxidatively-induced carbonylated proteins accompanies the increased functional severity of post-myocardial infarction heart failure in the setting of type 1 diabetes mellitus. Cardiovasc Pathol 22:473–480
Winterbourn CC (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 4:278–286
Mates JM, Segura JA, Alonso FJ, Marquez J (2012) Oxidative stress in apoptosis and cancer: an update. Arch Toxicol 86:1649–1665
Murphy MP (2012) Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal 16:476–495
Fridovich I (1995) Superoxide radical and superoxide dismutases. Annu Rev Biochem 64:97–112
Abreu IA, Cabelli DE (2010) Superoxide dismutases-a review of the metal-associated mechanistic variations. Biochim Biophys Acta 1804:263–274
Perry JJ, Shin DS, Getzoff ED, Tainer JA (2010) The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta 1804:245–262
Zelko IN, Mariani TJ, Folz RJ (2002) Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 33:337–349
Byrne JA, Grieve DJ, Cave AC, Shah AM (2003) Oxidative stress and heart failure. Arch Mal Coeur Vaiss 96:214–221
Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L et al (2005) CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24:367–380
Perez VI, Bokov A, Van Remmen H, Mele J, Ran Q et al (2009) Is the oxidative stress theory of aging dead? Biochim Biophys Acta 1790:1005–1014
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Majoor-Krakauer D, Willems PJ, Hofman A (2003) Genetic epidemiology of amyotrophic lateral sclerosis. Clin Genet 63:83–101
Andersen PM (2006) Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep 6:37–46
Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC et al (1995) Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11:376–381
Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L et al (1996) Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA 93:9782–9787
Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P et al (1998) A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet 18:159–163
Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B et al (1999) Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA 96:846–851
Pohjoismaki JL, Williams SL, Boettger T, Goffart S, Kim J et al (2013) Overexpression of Twinkle-helicase protects cardiomyocytes from genotoxic stress caused by reactive oxygen species. Proc Natl Acad Sci USA 110:19408–19413
Daosukho C, Ittarat W, Lin SM, Sawyer DB, Kiningham K et al (2005) Induction of manganese superoxide dismutase (MnSOD) mediates cardioprotective effect of tamoxifen (TAM). J Mol Cell Cardiol 39:792–803
Ohashi M, Runge MS, Faraci FM, Heistad DD (2006) MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 26:2331–2336
Sam F, Kerstetter DL, Pimental DR, Mulukutla S, Tabaee A et al (2005) Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail 11:473–480
Baumer AT, Flesch M, Wang X, Shen Q, Feuerstein GZ et al (2000) Antioxidative enzymes in human hearts with idiopathic dilated cardiomyopathy. J Mol Cell Cardiol 32:121–130
Dieterich S, Bieligk U, Beulich K, Hasenfuss G, Prestle J (2000) Gene expression of antioxidative enzymes in the human heart: increased expression of catalase in the end-stage failing heart. Circulation 101:33–39
Borchi E, Bargelli V, Stillitano F, Giordano C, Sebastiani M et al (2010) Enhanced ROS production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochim Biophys Acta 1802:331–338
Chelikani P, Fita I, Loewen PC (2004) Diversity of structures and properties among catalases. Cell Mol Life Sci 61:192–208
Kirkman HN, Gaetani GF (2007) Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem Sci 32:44–50
Nicholls P (2012) Classical catalase: ancient and modern. Arch Biochem Biophys 525:95–101
Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD et al (1991) Detection of catalase in rat heart mitochondria. J Biol Chem 266:22028–22034
Andreyev AY, Kushnareva YE, Starkov AA (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70:200–214
Cao C, Leng Y, Kufe D (2003) Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. J Biol Chem 278:29667–29675
Cao C, Leng Y, Li C, Kufe D (2003) Functional interaction between the c-Abl and Arg protein–tyrosine kinases in the oxidative stress response. J Biol Chem 278:12961–12967
Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE et al (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308:1909–1911
Treuting PM, Linford NJ, Knoblaugh SE, Emond MJ, Morton JF et al (2008) Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J Gerontol A Biol Sci Med Sci 63:813–822
Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ et al (2009) Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 119:2789–2797
Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ et al (2010) Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 9:536–544
Ho YS, Xiong Y, Ma W, Spector A, Ho DS (2004) Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem 279:32804–32812
Szabo C, Ischiropoulos H, Radi R (2007) Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6:662–680
Winterbourn CC, Hampton MB (2008) Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 45:549–561
Rhee SG, Woo HA, Kil IS, Bae SH (2012) Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem 287:4403–4410
Cao C, Leng Y, Huang W, Liu X, Kufe D (2003) Glutathione peroxidase 1 is regulated by the c-Abl and Arg tyrosine kinases. J Biol Chem 278:39609–39614
de Haan JB, Bladier C, Griffiths P, Kelner M, O’Shea RD et al (1998) Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J Biol Chem 273:22528–22536
Yoshida T, Maulik N, Engelman RM, Ho YS, Magnenat JL et al (1997) Glutathione peroxidase knockout mice are susceptible to myocardial ischemia reperfusion injury. Circulation 96:II-216–II-220
Torzewski M, Ochsenhirt V, Kleschyov AL, Oelze M, Daiber A et al (2007) Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 27:850–857
Brookes PS (2005) Mitochondrial H(+) leak and ROS generation: an odd couple. Free Radic Biol Med 38:12–23
Stowe DF, Camara AK (2009) Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid Redox Signal 11:1373–1414
Divakaruni AS, Brand MD (2011) The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 26:192–205
Palmieri F (2004) The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 447:689–709
Sluse FE (1996) Mitochondrial metabolite carrier family, topology, structure and functional properties: an overview. Acta Biochim Pol 43:349–360
el Moualij B, Duyckaerts C, Lamotte-Brasseur J, Sluse FE (1997) Phylogenetic classification of the mitochondrial carrier family of Saccharomyces cerevisiae. Yeast 13:573–581
Hughes J, Criscuolo F (2008) Evolutionary history of the UCP gene family: gene duplication and selection. BMC Evol Biol 8:306
Saito S, Saito CT, Shingai R (2008) Adaptive evolution of the uncoupling protein 1 gene contributed to the acquisition of novel nonshivering thermogenesis in ancestral eutherian mammals. Gene 408:37–44
Arechaga I, Ledesma A, Rial E (2001) The mitochondrial uncoupling protein UCP1: a gated pore. IUBMB Life 52:165–173
Modriansky M, Murdza-Inglis DL, Patel HV, Freeman KB, Garlid KD (1997) Identification by site-directed mutagenesis of three arginines in uncoupling protein that are essential for nucleotide binding and inhibition. J Biol Chem 272:24759–24762
Heaton GM, Wagenvoord RJ, Kemp A Jr, Nicholls DG (1978) Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation. Eur J Biochem 82:515–521
Nicholls DG, Bernson VS, Heaton GM (1978) The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Exp Suppl 32:89–93
Nicholls DG, Locke RM (1984) Thermogenic mechanisms in brown fat. Physiol Rev 64:1–64
Enerback S, Jacobsson A, Simpson EM, Guerra C, Yamashita H et al (1997) Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387:90–94
Krauss S, Zhang CY, Lowell BB (2005) The mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol 6:248–261
Cui Y, Xu X, Bi H, Zhu Q, Wu J et al (2006) Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: the role of reactive oxygen species in diabetic retinopathy. Exp Eye Res 83:807–816
Sale MM, Hsu FC, Palmer ND, Gordon CJ, Keene KL et al (2007) The uncoupling protein 1 gene, UCP1, is expressed in mammalian islet cells and associated with acute insulin response to glucose in African American families from the IRAS Family Study. BMC Endocr Disord 7:1
Fleury C, Neverova M, Collins S, Raimbault S, Champigny O et al (1997) Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet 15:269–272
Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E et al (2001) Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem 276:8705–8712
Azzu V, Affourtit C, Breen EP, Parker N, Brand MD (2008) Dynamic regulation of uncoupling protein 2 content in INS-1E insulinoma cells. Biochim Biophys Acta 1777:1378–1383
Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K (2004) Uncoupling proteins in human heart. Lancet 364:1786–1788
Sack MN (2006) Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res 72:210–219
Boss O, Samec S, Paoloni-Giacobino A, Rossier C, Dulloo A et al (1997) Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett 408:39–42
Vidal-Puig A, Solanes G, Grujic D, Flier JS, Lowell BB (1997) UCP3: an uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem Biophys Res Commun 235:79–82
Aguirre E, Cadenas S (2010) GDP and carboxyatractylate inhibit 4-hydroxynonenal-activated proton conductance to differing degrees in mitochondria from skeletal muscle and heart. Biochim Biophys Acta 1797:1716–1726
Harper ME, Himms-Hagen J (2001) Mitochondrial efficiency: lessons learned from transgenic mice. Biochim Biophys Acta 1504:159–172
Mailloux RJ, Harper ME (2012) Mitochondrial proticity and ROS signaling: lessons from the uncoupling proteins. Trends Endocrinol Metab 23:451–458
Brand MD, Esteves TC (2005) Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab 2:85–93
Mao W, Yu XX, Zhong A, Li W, Brush J et al (1999) UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett 443:326–330
Sanchis D, Fleury C, Chomiki N, Goubern M, Huang Q et al (1998) BMCP1, a novel mitochondrial carrier with high expression in the central nervous system of humans and rodents, and respiration uncoupling activity in recombinant yeast. J Biol Chem 273:34611–34615
Yu XX, Mao W, Zhong A, Schow P, Brush J et al (2000) Characterization of novel UCP5/BMCP1 isoforms and differential regulation of UCP4 and UCP5 expression through dietary or temperature manipulation. FASEB J 14:1611–1618
Alan L, Smolkova K, Kronusova E, Santorova J, Jezek P (2009) Absolute levels of transcripts for mitochondrial uncoupling proteins UCP2, UCP3, UCP4, and UCP5 show different patterns in rat and mice tissues. J Bioenerg Biomembr 41:71–78
Smorodchenko A, Rupprecht A, Sarilova I, Ninnemann O, Brauer AU et al (2009) Comparative analysis of uncoupling protein 4 distribution in various tissues under physiological conditions and during development. Biochim Biophys Acta 1788:2309–2319
Hanak P, Jezek P (2001) Mitochondrial uncoupling proteins and phylogenesis—UCP4 as the ancestral uncoupling protein. FEBS Lett 495:137–141
Ramsden DB, Ho PW, Ho JW, Liu HF, So DH et al (2012) Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav 2:468–478
Klingenberg M, Winkler E (1985) The reconstituted isolated uncoupling protein is a membrane potential driven H+ translocator. EMBO J 4:3087–3092
Winkler E, Klingenberg M (1994) Effect of fatty acids on H+ transport activity of the reconstituted uncoupling protein. J Biol Chem 269:2508–2515
Klingenberg M, Huang SG (1999) Structure and function of the uncoupling protein from brown adipose tissue. Biochim Biophys Acta 1415:271–296
Garlid KD, Orosz DE, Modriansky M, Vassanelli S, Jezek P (1996) On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. J Biol Chem 271:2615–2620
Rial E, Aguirregoitia E, Jimenez-Jimenez J, Ledesma A (2004) Alkylsulfonates activate the uncoupling protein UCP1: implications for the transport mechanism. Biochim Biophys Acta 1608:122–130
Shabalina IG, Jacobsson A, Cannon B, Nedergaard J (2004) Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem 279:38236–38248
Nicholls DG (2001) A history of UCP1. Biochem Soc Trans 29:751–755
Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ et al (2003) A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J 22:4103–4110
Murphy MP, Echtay KS, Blaikie FH, Asin-Cayuela J, Cocheme HM et al (2003) Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: studies using a mitochondria-targeted spin trap derived from α-phenyl-N-tert-butylnitrone. J Biol Chem 278:48534–48545
Esteves TC, Parker N, Brand MD (2006) Synergy of fatty acid and reactive alkenal activation of proton conductance through uncoupling protein 1 in mitochondria. Biochem J 395:619–628
Parker N, Affourtit C, Vidal-Puig A, Brand MD (2008) Energization-dependent endogenous activation of proton conductance in skeletal muscle mitochondria. Biochem J 412:131–139
Couplan E, del Mar Gonzalez-Barroso M, Alves-Guerra MC, Ricquier D, Goubern M et al (2002) No evidence for a basal, retinoic, or superoxide-induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J Biol Chem 277:26268–26275
Cannon B, Shabalina IG, Kramarova TV, Petrovic N, Nedergaard J (2006) Uncoupling proteins: a role in protection against reactive oxygen species—or not? Biochim Biophys Acta 1757:449–458
Nicholls DG (2006) The physiological regulation of uncoupling proteins. Biochim Biophys Acta 1757:459–466
Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S et al (2002) Superoxide activates mitochondrial uncoupling proteins. Nature 415:96–99
Considine MJ, Goodman M, Echtay KS, Laloi M, Whelan J et al (2003) Superoxide stimulates a proton leak in potato mitochondria that is related to the activity of uncoupling protein. J Biol Chem 278:22298–22302
Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S et al (2011) Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem 286:21865–21875
Mailloux RJ, Adjeitey CN, Xuan JY, Harper ME (2012) Crucial yet divergent roles of mitochondrial redox state in skeletal muscle vs. brown adipose tissue energetics. FASEB J 26:363–375
Feldmann HM, Golozoubova V, Cannon B, Nedergaard J (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab 9:203–209
Locke RM, Rial E, Scott ID, Nicholls DG (1982) Fatty acids as acute regulators of the proton conductance of hamster brown-fat mitochondria. Eur J Biochem 129:373–380
Robidoux J, Martin TL, Collins S (2004) Beta-adrenergic receptors and regulation of energy expenditure: a family affair. Annu Rev Pharmacol Toxicol 44:297–323
Cassard-Doulcier AM, Gelly C, Fox N, Schrementi J, Raimbault S et al (1993) Tissue-specific and beta-adrenergic regulation of the mitochondrial uncoupling protein gene: control by cis-acting elements in the 5′-flanking region. Mol Endocrinol 7:497–506
Kozak UC, Kopecky J, Teisinger J, Enerback S, Boyer B et al (1994) An upstream enhancer regulating brown-fat-specific expression of the mitochondrial uncoupling protein gene. Mol Cell Biol 14:59–67
Collins S, Cao W, Robidoux J (2004) Learning new tricks from old dogs: beta-adrenergic receptors teach new lessons on firing up adipose tissue metabolism. Mol Endocrinol 18:2123–2131
Patane G, Anello M, Piro S, Vigneri R, Purrello F et al (2002) Role of ATP production and uncoupling protein-2 in the insulin secretory defect induced by chronic exposure to high glucose or free fatty acids and effects of peroxisome proliferator-activated receptor-gamma inhibition. Diabetes 51:2749–2756
Tordjman K, Standley KN, Bernal-Mizrachi C, Leone TC, Coleman T et al (2002) PPARalpha suppresses insulin secretion and induces UCP2 in insulinoma cells. J Lipid Res 43:936–943
Takahashi A, Motomura K, Kato T, Yoshikawa T, Nakagawa Y et al (2005) Transgenic mice overexpressing nuclear SREBP-1c in pancreatic beta-cells. Diabetes 54:492–499
Affourtit C, Brand MD (2008) On the role of uncoupling protein-2 in pancreatic beta cells. Biochim Biophys Acta 1777:973–979
Oberkofler H, Hafner M, Felder T, Krempler F, Patsch W (2009) Transcriptional co-activator peroxisome proliferator-activated receptor (PPAR)gamma co-activator-1beta is involved in the regulation of glucose-stimulated insulin secretion in INS-1E cells. J Mol Med (Berl) 87:299–306
Li LX, Skorpen F, Egeberg K, Jorgensen IH, Grill V (2002) Induction of uncoupling protein 2 mRNA in beta-cells is stimulated by oxidation of fatty acids but not by nutrient oversupply. Endocrinology 143:1371–1377
Li LX, Skorpen F, Egeberg K, Jorgensen IH, Grill V (2001) Uncoupling protein-2 participates in cellular defense against oxidative stress in clonal beta-cells. Biochem Biophys Res Commun 282:273–277
Giardina TM, Steer JH, Lo SZ, Joyce DA (2008) Uncoupling protein-2 accumulates rapidly in the inner mitochondrial membrane during mitochondrial reactive oxygen stress in macrophages. Biochim Biophys Acta 1777:118–129
Samec S, Seydoux J, Dulloo AG (1998) Interorgan signaling between adipose tissue metabolism and skeletal muscle uncoupling protein homologs: is there a role for circulating free fatty acids? Diabetes 47:1693–1698
Solanes G, Pedraza N, Iglesias R, Giralt M, Villarroya F (2003) Functional relationship between MyoD and peroxisome proliferator-activated receptor-dependent regulatory pathways in the control of the human uncoupling protein-3 gene transcription. Mol Endocrinol 17:1944–1958
Lanni A, Beneduce L, Lombardi A, Moreno M, Boss O et al (1999) Expression of uncoupling protein-3 and mitochondrial activity in the transition from hypothyroid to hyperthyroid state in rat skeletal muscle. FEBS Lett 444:250–254
Solanes G, Pedraza N, Iglesias R, Giralt M, Villarroya F (2000) The human uncoupling protein-3 gene promoter requires MyoD and is induced by retinoic acid in muscle cells. FASEB J 14:2141–2143
Busquets S, Sanchis D, Alvarez B, Ricquier D, Lopez-Soriano FJ et al (1998) In the rat, tumor necrosis factor alpha administration results in an increase in both UCP2 and UCP3 mRNAs in skeletal muscle: a possible mechanism for cytokine-induced thermogenesis? FEBS Lett 440:348–350
Boss O, Samec S, Kuhne F, Bijlenga P, Assimacopoulos-Jeannet F et al (1998) Uncoupling protein-3 expression in rodent skeletal muscle is modulated by food intake but not by changes in environmental temperature. J Biol Chem 273:5–8
Cadenas S, Buckingham JA, Samec S, Seydoux J, Din N et al (1999) UCP2 and UCP3 rise in starved rat skeletal muscle but mitochondrial proton conductance is unchanged. FEBS Lett 462:257–260
Bordone L, Motta MC, Picard F, Robinson A, Jhala US et al (2006) Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol 4:e31
Amat R, Solanes G, Giralt M, Villarroya F (2007) SIRT1 is involved in glucocorticoid-mediated control of uncoupling protein-3 gene transcription. J Biol Chem 282:34066–34076
Hurtaud C, Gelly C, Bouillaud F, Levi-Meyrueis C (2006) Translation control of UCP2 synthesis by the upstream open reading frame. Cell Mol Life Sci 63:1780–1789
Hurtaud C, Gelly C, Chen Z, Levi-Meyrueis C, Bouillaud F (2007) Glutamine stimulates translation of uncoupling protein 2mRNA. Cell Mol Life Sci 64:1853–1860
Puigserver P, Herron D, Gianotti M, Palou A, Cannon B et al (1992) Induction and degradation of the uncoupling protein thermogenin in brown adipocytes in vitro and in vivo. Evidence for a rapidly degradable pool. Biochem J 284(Pt 2):393–398
Moazed B, Desautels M (2002) Control of proteolysis by norepinephrine and insulin in brown adipocytes: role of ATP, phosphatidylinositol 3-kinase, and p70 S6K. Can J Physiol Pharmacol 80:541–552
Moazed B, Desautels M (2002) Differentiation-dependent expression of cathepsin D and importance of lysosomal proteolysis in the degradation of UCP1 in brown adipocytes. Can J Physiol Pharmacol 80:515–525
Rousset S, Mozo J, Dujardin G, Emre Y, Masscheleyn S et al (2007) UCP2 is a mitochondrial transporter with an unusual very short half-life. FEBS Lett 581:479–482
Azzu V, Mookerjee SA, Brand MD (2010) Rapid turnover of mitochondrial uncoupling protein 3. Biochem J 426:13–17
Azzu V, Brand MD (2010) Degradation of an intramitochondrial protein by the cytosolic proteasome. J Cell Sci 123:578–585
Kitiphongspattana K, Mathews CE, Leiter EH, Gaskins HR (2005) Proteasome inhibition alters glucose-stimulated (pro)insulin secretion and turnover in pancreatic {beta}-cells. J Biol Chem 280:15727–15734
Yan FF, Lin CW, Cartier EA, Shyng SL (2005) Role of ubiquitin-proteasome degradation pathway in biogenesis efficiency of {beta}-cell ATP-sensitive potassium channels. Am J Physiol Cell Physiol 289:C1351–C1359
Kawaguchi M, Minami K, Nagashima K, Seino S (2006) Essential role of ubiquitin-proteasome system in normal regulation of insulin secretion. J Biol Chem 281:13015–13020
Sasahara M, Nishi M, Kawashima H, Ueda K, Sakagashira S et al (2004) Uncoupling protein 2 promoter polymorphism -866G/A affects its expression in beta-cells and modulates clinical profiles of Japanese type 2 diabetic patients. Diabetes 53:482–485
Jia JJ, Zhang X, Ge CR, Jois M (2009) The polymorphisms of UCP2 and UCP3 genes associated with fat metabolism, obesity and diabetes. Obes Rev 10:519–526
Dalgaard LT (2011) Genetic variance in uncoupling protein 2 in relation to obesity, type 2 diabetes, and related metabolic traits: focus on the functional -866G>A promoter variant (rs659366). J Obes 2011:340241
Souza BM, Assmann TS, Kliemann LM, Gross JL, Canani LH et al (2011) The role of uncoupling protein 2 (UCP2) on the development of type 2 diabetes mellitus and its chronic complications. Arq Bras Endocrinol Metabol 55:239–248
Liu J, Li J, Li WJ, Wang CM (2013) The role of uncoupling proteins in diabetes mellitus. J Diabetes Res 2013:585897
Boss O, Hagen T, Lowell BB (2000) Uncoupling proteins 2 and 3: potential regulators of mitochondrial energy metabolism. Diabetes 49:143–156
Nedergaard J, Cannon B (2003) The ‘novel’ ‘uncoupling’ proteins UCP2 and UCP3: What do they really do? Pros and cons for suggested functions. Exp Physiol 88:65–84
Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME et al (2005) Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112:2686–2695
Hoeks J, Hesselink MK, van Bilsen M, Schaart G, van der Vusse GJ et al (2003) Differential response of UCP3 to medium versus long chain triacylglycerols; manifestation of a functional adaptation. FEBS Lett 555:631–637
Laskowski KR, Russell RR 3rd (2008) Uncoupling proteins in heart failure. Curr Heart Fail Rep 5:75–79
Nabben M, Hoeks J (2008) Mitochondrial uncoupling protein 3 and its role in cardiac- and skeletal muscle metabolism. Physiol Behav 94:259–269
Van der Lee KA, Willemsen PH, Samec S, Seydoux J, Dulloo AG et al (2001) Fasting-induced changes in the expression of genes controlling substrate metabolism in the rat heart. J Lipid Res 42:1752–1758
Young ME, Patil S, Ying J, Depre C, Ahuja HS et al (2001) Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. FASEB J 15:833–845
Lee CK, Allison DB, Brand J, Weindruch R, Prolla TA (2002) Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc Natl Acad Sci USA 99:14988–14993
Kahaly GJ, Dillmann WH (2005) Thyroid hormone action in the heart. Endocr Rev 26:704–728
Jekabsons MB, Gregoire FM, Schonfeld-Warden NA, Warden CH, Horwitz BA (1999) T(3) stimulates resting metabolism and UCP-2 and UCP-3 mRNA but not nonphosphorylating mitochondrial respiration in mice. Am J Physiol 277:E380–E389
Barbe P, Larrouy D, Boulanger C, Chevillotte E, Viguerie N et al (2001) Triiodothyronine-mediated up-regulation of UCP2 and UCP3 mRNA expression in human skeletal muscle without coordinated induction of mitochondrial respiratory chain genes. FASEB J 15:13–15
Lanni A, Moreno M, Lombardi A, Goglia F (2003) Thyroid hormone and uncoupling proteins. FEBS Lett 543:5–10
Boehm EA, Jones BE, Radda GK, Veech RL, Clarke K (2001) Increased uncoupling proteins and decreased efficiency in palmitate-perfused hyperthyroid rat heart. Am J Physiol Heart Circ Physiol 280:H977–H983
Short KR, Nygren J, Barazzoni R, Levine J, Nair KS (2001) T(3) increases mitochondrial ATP production in oxidative muscle despite increased expression of UCP2 and -3. Am J Physiol Endocrinol Metab 280:E761–E769
Taegtmeyer H, Razeghi P, Young ME (2002) Mitochondrial proteins in hypertrophy and atrophy: a transcript analysis in rat heart. Clin Exp Pharmacol Physiol 29:346–350
Young ME, Laws FA, Goodwin GW, Taegtmeyer H (2001) Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J Biol Chem 276:44390–44395
Teshima Y, Akao M, Jones SP, Marban E (2003) Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res 93:192–200
McLeod CJ, Aziz A, Hoyt RF Jr, McCoy JP Jr, Sack MN (2005) Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. J Biol Chem 280:33470–33476
Bodyak N, Rigor DL, Chen YS, Han Y, Bisping E et al (2007) Uncoupling protein 2 modulates cell viability in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol 293:H829–H835
Cabrera JA, Ziemba EA, Colbert R, Kelly RF, Kuskowski M et al (2012) Uncoupling protein-2 expression and effects on mitochondrial membrane potential and oxidant stress in heart tissue. Transl Res 159:383–390
Ozcan C, Palmeri M, Horvath TL, Russell KS, Russell RR 3rd (2013) Role of uncoupling protein 3 in ischemia-reperfusion injury, arrhythmias, and preconditioning. Am J Physiol Heart Circ Physiol 304:H1192–H1200
Perrino C, Schiattarella GG, Sannino A, Pironti G, Petretta MP et al (2013) Genetic deletion of uncoupling protein 3 exaggerates apoptotic cell death in the ischemic heart leading to heart failure. J Am Heart Assoc 2:e000086
Noma T, Nishiyama A, Mizushige K, Murakami K, Tsuji T et al (2001) Possible role of uncoupling protein in regulation of myocardial energy metabolism in aortic regurgitation model rats. FASEB J 15:1206–1208
Murakami K, Mizushige K, Noma T, Tsuji T, Kimura S et al (2002) Perindopril effect on uncoupling protein and energy metabolism in failing rat hearts. Hypertension 40:251–255
Bugger H, Guzman C, Zechner C, Palmeri M, Russell KS et al (2011) Uncoupling protein downregulation in doxorubicin-induced heart failure improves mitochondrial coupling but increases reactive oxygen species generation. Cancer Chemother Pharmacol 67:1381–1388
Murray AJ, Cole MA, Lygate CA, Carr CA, Stuckey DJ et al (2008) Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J Mol Cell Cardiol 44:694–700
Safari F, Bayat G, Shekarforoush S, Hekmatimoghaddam S, Anvari Z et al (2013) Expressional profile of cardiac uncoupling protein-2 following myocardial ischemia reperfusion in losartan- and ramiprilat-treated rats. J Renin Angiotensin Aldosterone Syst. http://jra.sagepub.com/content/early/2013/01/31/1470320312474050.full.pdf+html
Safari F, Anvari Z, Moshtaghioun S, Javan M, Bayat G et al (2014) Differential expression of cardiac uncoupling proteins 2 and 3 in response to myocardial ischemia-reperfusion in rats. Life Sci 98:68–74
McFalls EO, Sluiter W, Schoonderwoerd K, Manintveld OC, Lamers JM et al (2006) Mitochondrial adaptations within chronically ischemic swine myocardium. J Mol Cell Cardiol 41:980–988
Almsherqi ZA, McLachlan CS, Slocinska MB, Sluse FE, Navet R et al (2006) Reduced cardiac output is associated with decreased mitochondrial efficiency in the non-ischemic ventricular wall of the acute myocardial-infarcted dog. Cell Res 16:297–305
Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH et al (2001) Metabolic gene expression in fetal and failing human heart. Circulation 104:2923–2931
Razeghi P, Young ME, Ying J, Depre C, Uray IP et al (2002) Downregulation of metabolic gene expression in failing human heart before and after mechanical unloading. Cardiology 97:203–209
Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD (2011) Targeting fatty acid and carbohydrate oxidation–a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta 1813:1333–1350
Vettor R, Fabris R, Serra R, Lombardi AM, Tonello C et al (2002) Changes in FAT/CD36, UCP2, UCP3 and GLUT4 gene expression during lipid infusion in rat skeletal and heart muscle. Int J Obes Relat Metab Disord 26:838–847
Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR et al (2003) Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res 92:518–524
Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K (2005) Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes 54:3496–3502
Ray J, Noll F, Daut J, Hanley PJ (2002) Long-chain fatty acids increase basal metabolism and depolarize mitochondria in cardiac muscle cells. Am J Physiol Heart Circ Physiol 282:H1495–H1501
Garvey WT, Hardin D, Juhaszova M, Dominguez JH (1993) Effects of diabetes on myocardial glucose transport system in rats: implications for diabetic cardiomyopathy. Am J Physiol 264:H837–H844
Stanley WC, Hall JL, Hacker TA, Hernandez LA, Whitesell LF (1997) Decreased myocardial glucose uptake during ischemia in diabetic swine. Metabolism 46:168–172
Hidaka S, Kakuma T, Yoshimatsu H, Sakino H, Fukuchi S et al (1999) Streptozotocin treatment upregulates uncoupling protein 3 expression in the rat heart. Diabetes 48:430–435
Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE et al (2003) Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation 107:3040–3046
Gerber LK, Aronow BJ, Matlib MA (2006) Activation of a novel long-chain free fatty acid generation and export system in mitochondria of diabetic rat hearts. Am J Physiol Cell Physiol 291:C1198–C1207
Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357:1121–1135
Kalogeris T, Baines CP, Krenz M, Korthuis RJ (2012) Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298:229–317
Ishioka K, Kanehira K, Sasaki N, Kitamura H, Kimura K et al (2002) Canine mitochondrial uncoupling proteins: structure and mRNA expression of three isoforms in adult beagles. Comp Biochem Physiol B: Biochem Mol Biol 131:483–489
Yellon DM, Downey JM (2003) Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev 83:1113–1151
Sanada S, Komuro I, Kitakaze M (2011) Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol 301:H1723–H1741
Bell RM, Yellon DM (2012) Conditioning the whole heart–not just the cardiomyocyte. J Mol Cell Cardiol 53:24–32
Hausenloy DJ (2013) Cardioprotection techniques: preconditioning, postconditioning and remote conditioning (basic science). Curr Pharm Des 19:4544–4563
Brooks MJ, Andrews DT (2013) Molecular mechanisms of ischemic conditioning: translation into patient outcomes. Future Cardiol 9:549–568
Diano S, Matthews RT, Patrylo P, Yang L, Beal MF et al (2003) Uncoupling protein 2 prevents neuronal death including that occurring during seizures: a mechanism for preconditioning. Endocrinology 144:5014–5021
Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G et al (2003) Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med 9:1062–1068
Miller TD, Christian TF, Hopfenspirger MR, Hodge DO, Gersh BJ et al (1995) Infarct size after acute myocardial infarction measured by quantitative tomographic 99mTc sestamibi imaging predicts subsequent mortality. Circulation 92:334–341
Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W et al (1997) Apoptosis in the failing human heart. N Engl J Med 336:1131–1141
Bienengraeber M, Ozcan C, Terzic A (2003) Stable transfection of UCP1 confers resistance to hypoxia/reoxygenation in a heart-derived cell line. J Mol Cell Cardiol 35:861–865
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529
Bers DM (2008) Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70:23–49
Kranias EG, Hajjar RJ (2012) Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 110:1646–1660
Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF (2007) Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2 + uniport. Nat Cell Biol 9:445–452
Brookes PS, Parker N, Buckingham JA, Vidal-Puig A, Halestrap AP et al (2008) UCPs—unlikely calcium porters. Nat Cell Biol 10:1235–1237; author reply 1237–1240
Turner JD, Gaspers LD, Wang G, Thomas AP (2010) Uncoupling protein-2 modulates myocardial excitation–contraction coupling. Circ Res 106:730–738
Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P et al (2003) Protective role of uncoupling protein 2 in atherosclerosis. Circulation 107:388–390
Oberkofler H, Iglseder B, Klein K, Unger J, Haltmayer M et al (2005) Associations of the UCP2 gene locus with asymptomatic carotid atherosclerosis in middle-aged women. Arterioscler Thromb Vasc Biol 25:604–610
Conflict of interest
This manuscript has not been, nor will be published elsewhere, has been read and is submitted with the approval of all authors, all of which participated in the writing of the manuscript with no conflict of interest in its publication in Heart Failure Reviews.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Akhmedov, A.T., Rybin, V. & Marín-García, J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail Rev 20, 227–249 (2015). https://doi.org/10.1007/s10741-014-9457-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-014-9457-4