
Abstract
Renal dysfunction is often present and/or worsens in patients with heart failure and this is associated with increased costs of care, complications and mortality. The cardiorenal syndrome can be defined as the presence or development of renal dysfunction in patients with heart failure. Its mechanisms are likely related to low cardiac output, increased venous congestion and renal venous pressure, neurohormonal and inflammatory activation and local changes, such as adenosine release. Many drugs, including loop diuretics, may contribute to worsening renal function through the activation of some of these mechanisms. Renal damage is conventionally defined by the increase in creatinine and blood urea nitrogen blood levels. However, these changes may be not related with renal injury or prognosis. New biomarkers of renal injury seem promising but still need to be validated. Thus, despite the epidemiological evidence, we are still lacking of satisfactory tools to assess renal injury and function and its prognostic significance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute heart failure (AHF) is the most important cause of hospitalization in the adult subjects in Western countries with approximately 3 million hospitalizations as first diagnosis or contributing factor in United States [1, 2] and similar rates have been described in Europe [3]. Although the optimization of therapy and the introduction of new drugs and devices have improved outcomes, the prognosis of the patients remains unsatisfactory. Mortality rates are still high with in-hospital mortality rates of 3–4%, post-discharge mortality rates of approximately 10% and rehospitalization rates of about 25% in the 60–90 days after an admission for AHF [4, 5]. Renal dysfunction seems to have a major impact both on patients’ clinical presentation and treatment as well as on their prognosis [6–8]. Studies have been conducted mainly based on an assessment of renal function using serum creatinine levels and their changes during the AHF hospitalization. We will focus in the present article on the results obtained with the assessment of renal function through serum creatinine levels and the clinical significance of these findings.
Epidemiology of renal dysfunction in heart failure
The prevalence of renal dysfunction is steadily increasing. In United States, it is estimated that 6.2 million people have serum creatinine levels (sCr) >1.5 mg/dl [9]. Renal dysfunction is one of the most common comorbidity among patients hospitalized for heart failure (HF). In the Acute Decompensated Heart Failure National Registry (ADHERE), 30% of the patients had renal failure with 21% with sCr >2.0 mg/dl [6]. Similar data have been described in other studies. In the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE), the proportion of patients with an estimated glomerular filtration rate (eGFR) <60 ml/min was 31.4% [10]. Among patients with chronic heart failure, in the Digitalis Investigation Group (DIG) trial, the prevalence of renal dysfunction was 45% [11]. The prevalence of moderate to severe renal dysfunction was 36% in the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) [12] and 33.4% in the recent the Eplerenone in Mild Patients Hospitalization And Survival Study in Heart Failure (EMPHASIS-HF) [13]. Renal dysfunction has been associated with poorer outcomes in patients with HF with higher in-hospital, post-discharge and long-term mortality and prolonged duration of the hospitalization [14]. In a meta-analysis, annual mortality rates were 26% in patients without renal dysfunction versus 41% in the patients with any impairment of renal function and 51% (P < 0.001) in those with moderate to severe impairment (P < 0.001). Subgroups analysis showed that patients with renal dysfunction, symptoms of congestion and NYHA functional class III or IV had a higher mortality risk [15].
Patients hospitalized for AHF often develop worsening renal function. Although still controversial, worsening renal function is often defined as an increase in sCr ≥0.3 mg/dl from baseline value [15–17]. In a first meta-analysis of 16 studies including 80,098 patients, worsening renal function was associated with a 47% increase in one-year mortality, with a 33% increase in mortality for every 1 mg/dl increase in sCr, whereas its relationship with rehospitalizations was marginal [15]. Damman et al. reported a 61% increase in risk of death and a 30% increase in the risk of all-cause readmission associated with worsening renal function after 2–6 months of follow-up [18].
Causes of renal dysfunction in AHF
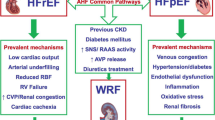
There is a close relationship between renal and cardiac dysfunction and this has been the basis of the concept of cardio-renal syndrome (CRS) (Fig. 1). However, it is not yet clear how the biochemical, hormonal and hemodynamic pathophysiological factors and pharmacological interventions interact to cause this syndrome (Table 1).

Mechanisms of cardiorenal syndrome. AVP Arginine vasopressin
Neurohormonal activation
Renin-angiotensin-aldosterone system
The renin-angiotensin-aldosterone system (RAAS) causes sodium retention, ventricular remodeling and determinates poor outcomes of the patients with HF. ACE inhibitors and angiotensin receptor blockers (ARBs) protect renal function in patients with hypertension and diabetes AT II has untoward effects on kidney function likely mediated by endothelial dysfunction, oxidative stress, inflammation and fibrosis [19–21].
Sympathetic activity
Although the effects of hyperactivity of the sympathetic nervous system on the heart are well described, little is known about its renal effects. In the kidneys, increase in sympathetic activity leads to activation of the RAAS, and then further sodium retention and baroreceptors-mediated renal vasoconstriction [22]. These effects are greater in patients with HF and advanced renal dysfunction due to a lower clearance of circulating catecholamines. In a recent pilot study in patients with resistant hypertension who underwent renal sympathetic denervation, investigators have observed a significant improvement in estimated glomerular filtration rate (eGFR) in 24% of cases [23].
Cardio-renal anemia syndrome
Anemia is often observed in patients with HF and renal dysfunction [24, 25]. It has multiple mechanisms including erythropoietin deficiency (in advanced renal dysfunction), insensitivity to elevated erythropoietin levels caused by inflammation and iron deficiency [26]. Erythropoietin has antiapoptotic and antioxidant effects, independent from its effects on serum hemoglobin levels [27, 28]. It is not clear whether anemia is a mere marker of worse prognosis in HF or an active mechanism contributing to CRS.
Low cardiac output
Patients with HF may progress to a chronic low cardiac output state with renal hypoperfusion and reduced eGFR [29]. This causes activation of renin release by the juxtaglomerular cells located in the afferent arterioles with consequent RAAS activation leading to sodium retention, volume expansion and ventricular remodeling [30].
Central venous pressure and intra-abdominal pressure elevation
An increase in central venous pressure, as in HF patients, causes an increase in the glomerular efferent arteriole pressure with a reduction of the glomerular filtration pressure gradient and a fall of the glomerular filtration rate. In a study, 145 consecutive patients admitted for AHF and treated with intensive medical therapy guided by pulmonary artery catheterization, central venous pressure either on admission or after intensive medical therapy, was the most important determinant of the development of worsening renal function, defined as an increase in sCr from admission of ≥0.3 mg/dl. No difference in the other hemodynamic variables, including blood pressure, pulmonary artery pressure and pulmonary wedge pressure were found between the patients who developed or not worsening renal function. Rather unexpectedly, cardiac index was greater in the patients with worsening renal function both at baseline and after treatment [31]. Consistent results were simultaneously published by Damman et al. who studied the relationship between renal dysfunction and hemodynamic parameters in 2,557 patients hospitalized for cardiac catheterization. They found that central venous pressure was the most important determinant of renal dysfunction and was the most important independent predictor of mortality [32]. In another study in 40 consecutive patients admitted for acutely decompensated HF, intra-abdominal pressure, measured by a transvescical technique, was higher in the patients with worse renal function and, importantly, a significant correlation was observed between reduction in intra-abdominal pressure and improved renal function after treatment in patients with elevated values at baseline [33]. Further studies have confirmed the role of central venous pressure as a determinant of renal dysfunction in patients with HF [34, 35].
Intrarenal mechanisms—adenosine release
Adenosine concentration is increased in patients with HF [36]. In the kidney, adenosine is released by the juxtaglomerular cells in response to increase in sodium load in the distal tubule, sensed by the macula densa cells. Adenosine binds to A1-receptors located in the proximal tubule and afferent arterioles of the glomerulus. This leads to a reduction in intracellular cyclic adenosine monophosphate (cAMP) and an increase the activity of basolateral Na+-HCO3 − symporter in the proximal tubule and to constriction of the afferent glomerular arteriole. Thus, adenosine release following an increase of sodium load to the distal tubule, as during intensive diuretic treatment for AHF, leads to sodium retention and reduces GFR. Thus adenosine release may be a major mechanism of renal dysfunction after high-dose furosemide treatment [37]. However, studies with type 1a-adenosine receptors blocking agents have failed to show a major improvement in renal function despite mild diuretic and saluretic effects, likely related to the effects on the proximal tubule, were found [38].
Effects of treatment on serum creatinine levels
Many drugs used in treatment of AHF can directly or indirectly affect renal function. Loop diuretics are the mainstay treatment of AHF. However, loop diuretics, mainly furosemide, have been associated with increase in sCr levels and worse outcomes in several series of patients hospitalized for heart failure and in outpatients setting [39–42]. Moreover, patients may become resistant to their effects so that higher furosemide doses may be necessary to achieve a satisfactory diuretic and saluretic response with, in more advanced stages, the need of further tools, such as hemodialysis or peritoneal dialysis to treat fluid overload. The mechanisms of diuretic resistance are multiple and hence treatment may vary. They have been thoroughly examined in recent reviews [43–45] and are outlined in Table 2.
In a sub-analysis of the SOLVD (Studies Of Left Ventricular Dysfunction), the use of loop diuretics was a predictor of increased risk of renal impairment in the multivariable model [35]. In the ESCAPE trial, the investigators found a small correlation between maximal diuretic dose and changes in serum creatinine (r = 0.043; P = 0.412) and eGFR (r = −0.0149; P = 0.777) [46]. In a study conducted at our center on 318 consecutive patients admitted for AHF, worsening renal function, defined by an increase in sCr of both ≥0.3 mg/dl and ≥25% from the values on admission, was found in 107 patients (34%) [40]. The patients who had developed worsening renal function were more likely have a history of preexisting renal disease and more severe HF symptoms, at the time of admission. In addition, they were treated with higher doses of furosemide during the hospitalization and at discharge. At multivariable analysis, the only independent predictors of worsening renal function were history of chronic kidney disease, furosemide daily dose on admission, New York Heart Association (NYHA) functional class and left ventricular ejection fraction. Thus, this study suggests that furosemide administration may be independently related with the development of renal dysfunction, defined based on sCr levels.
In the recent Determining Optimal Dose and Duration of Diuretic Treatment in People with Acute Heart Failure (DOSE-AHF) study, patients who received high doses of furosemide, compared to patients who received lower doses, had a greater rate of worsening renal function, but similar prognosis at 60 days [47]. These last data suggest that an increase in diuretic dose may actually have beneficial effects when used in patients who need them because of fluid overload.
Thus, overdosing of diuretic therapy can cause fluid contraction and arterial underfilling with further neurohormonal activation and HF progression. However, also diuretic underdosing may be detrimental as it may cause persistent fluid overload and congestion with persistent symptoms, further deterioration in renal function, through the effects of increased renal venous pressure, and neurohormonal activation through the effects of increased myocardial stress.
Other agents may have an impact on renal function. Administration of inotropic agents may be indicated to improve left ventricular systolic function and, hence, renal perfusion and function and the response to diuretic therapy. Dopamine, when administered at low doses, may rather selectively improve renal blood flow in both the large conductance and small resistance renal blood vessels through its action on DA1 receptors and this was attended by an improvement in diuresis and by small, but favorable, changes in renal function, in some studies [48–50]. Despite these results, no data have shown favorable effects of dopamine infusion on major outcomes, defined as either mortality, rehospitalizations and prevention of renal damage [51]. Thus, there is no evidence to recommend dopamine administration for the protection of renal function in patients with fluid overload and need of diuretic treatment. In the recent Dopamine in Acute Decompensated Heart Failure (DAD-HF) trial, 60 consecutive patients hospitalized for AHF were randomized to high doses of furosemide or low doses of furosemide plus low doses of dopamine. Worsening renal function and hypokaliemia were more frequent in the high doses arm (P = 0.042 and P = 0.003, respectively), although the 60 days outcomes were similar in both groups. The study had some limitations, related to its small sample size, which do not allow to draw conclusions regarding the effects on outcomes, as well as with regards of the relatively high average systolic blood pressure on admission (176 ± 33 and 157 ± 28 mmHg in high-dose furosemide and dopamine group, respectively) [52]. Lastly, a low furosemide dose regimen was not examined. For these reasons, further studies are needed.
Dobutamine administration has been associated with an increase in diuresis and natriuresis, likely caused by the increase in cardiac output, as renal blood flow was not selectively increased [53]. Levosimendan was studied in 88 patients admitted for AHF requiring inotropic therapy. Levosimendan administration was associated with an improvement from baseline in eGFR, compared with dobutamine, at 24 (+15%, P < 0.001) and 72 hours (45%, P < 0.001). Nevertheless, these data were obtained in small, single-center trials and need confirmation by larger studies.
The beneficial effects of natriuretic peptides explain the active research for synthetic analogs to be used in the treatment of HF [54]. Among them, nesiritide, a recombinant human BNP, is the most important, as studied in large randomized, controlled, trials [55, 56] and approved for treatment in the United States and other countries but not in most of the European countries [57, 58]. It has been associated with an improvement in dyspnea and a reduction in pulmonary capillary wedge pressure, compared with placebo, in randomized studies [28]. BNP infusion has been shown to increase diuresis and natriuresis in normal subjects and to prevent deterioration of renal function in post-cardiac surgery patients [59]. However, these effects were not shown in patients with HF [60]. Meta-analyses of previous randomized trials had raised concerns regarding untoward effects on renal function and mortality of nesiritide infusion [61]. In order to assess the effects of nesiritide on symptoms and outcomes of the patients with acute HF, the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF), was designed [56]. This study included 7,141 patients with AHF, randomized to placebo or nesiritide. The trial confirmed that nesiritide administration was associated with an improvement in dyspnea, compared with placebo, although not meeting the prespecified criteria for statistical significance. No effects on outcomes were found. Thus, ASCEND-AHF showed the safety of nesiritide administration although with only mild effects on symptoms and no effects on outcomes [62]. Carperitide (recombinant ANP), urodilatin and ularitide are other natriuretic peptides used in some countries (carperitide, available in Japan) and/or undergoing clinical research [63].
Adenosine type A1 antagonists: a renal specific therapy?
Adenosine acts on receptors expressed in the afferent arteriole, causing vasoconstriction and reduced renal blood flow. It also promotes sodium retention and activation of the tubuloglomerular feedback, a mechanism through which the increased sodium load in the tubule leads to increased adenosine release and hence afferent arteriole constriction and reduced eGFR [64].
Blockade of adenosine type 1 receptors was therefore a mechanism potentially useful for renal protection. The first small studies with A1 adenosine receptor antagonists were consistent with this hypothesis. Gottlieb et al. studied 12 patients with HF receiving either BG9719 alone, furosemide alone or their combination. The administration of the adenosine antagonist was associated with a lower decrease in eGFR after concomitant furosemide administration (P < 0.005) with a further increase in renal sodium excretion. Further studies confirmed these favorable findings [65, 66].
Unfortunately, as often in clinical cardiology, the results of smaller, mechanistic trials were not translated in larger multicenter trial. In the PROTECT trial (A Placebo-controlled Randomized study of the selective A1 adenosine receptor antagonist rolofylline for patients hospitalized with acute heart failure and volume Overload to assess Treatment Effect on Congestion and renal function), 2,033 patients admitted for AHF were enrolled and randomized 2:1 to the type 1A adenosine antagonist rolofylline or placebo. Worsening renal function, defined as an increase from baseline ≥0.3 mg/dl of serum creatinine at day 7 from enrollment persisting at day 14 was, for the first time, included as a component of the primary end-point and as an essential component, together with dialysis or hemofiltration, of a secondary end-point. However, despite the favorable results of preliminary studies, PROTECT failed to show any beneficial effect of rolofylline on worsening renal dysfunction. Actually, the proportion of patients who developed worsening renal function was numerically greater with rolofylline compared with placebo, whereas rolofylline, likely through its mild diuretic effects, had a favorable effect on dyspnea as well as short-term mortality [67]. Rolofylline was also associated with higher rates of seizures and stroke, compared with placebo, and this has further inhibited any development of the drug by the sponsoring company [68].
In conclusion, many drugs can affect serum creatinine levels despite small effects on outcomes. This is consistent with limitations in our current assessment of renal function, mainly through serum creatinine changes. We will discuss herewith limitations of serum creatinine measurements as well as new tools for assessing renal function.
Is serum creatinine an accurate marker of renal function and renal injury?
Definitions of renal dysfunction are almost universally based on serum creatinine changes. However, many limitations of serum creatinine, as a marker of renal dysfunction, have been recently shown and up to 20 or more markers of renal dysfunction are currently in an advanced stage of assessment (Table 3) [69]. Serum creatinine is related to other variables, namely, age, gender and muscle mass [70, 71]. A subject with increased muscle mass has higher serum creatinine levels, compared with a cachectic patient and this is important as HF, as many chronic diseases, associated with muscle wasting especially in more advanced stages.
Serum creatinine is not sensitive for the detection of renal injury, above all in experimental models or subjects with a substantial renal reserve. Histological studies have shown that a relatively large amount of renal damage can occur without producing a change in eGFR calculated on the basis of serum creatinine levels. The percentage changes in serum creatinine after severe acute renal injury are highly dependent on baseline kidney function so that serum creatinine levels start to increase only at advanced stages of renal dysfunction when renal function is initially normal whereas they overestimate renal damage when renal function is already abnormal so that a relatively large increase in serum creatinine may occur with a slighter decrease in renal function [71–74]. Because of this exponential relationship between serum creatinine changes and eGFR changes, worsening renal function may be better defined by either an absolute increase from baseline and a percent increase. We recently showed that worsening renal function was related with outcomes, when expressed as both a ≥0.3 mg/dl increase and ≥25% increase from baseline values, whereas it had no independent prognostic value, when defined only by absolute changes from baseline [40].
Moreover, serum creatinine has a slow kinetics. Large changes in eGFR may be associated with relatively small changes in serum creatinine levels in the first 24–48 h after acute kidney injury resulting in a delayed diagnosis and underestimation of the degree of injury. Serum creatinine kinetics is also dependent on baseline renal function so that time to reach a 50% increase in serum creatinine ranges from 4 h when renal function is normal at baseline to up to 27 h in stages of advanced renal dysfunction [75].
Serum creatinine levels measure renal function but not renal injury. This has another shortcoming as changes in renal function may occur as a consequence of changes in volume status in the absence of any renal damage. This often occurs in patients undergoing treatment with relatively high doses of furosemide for acute HF. In an analysis of the ESCAPE trial, Testani et al. studied the correlation between hemoconcentration and renal function. Hemoconcentration was defined on the basis of change in the hematocrit, serum albumin and total protein levels, and worsening renal function was defined by a ≥20% decrease in GFR. Hemoconcentration was strongly associated with worsening renal function (odds ratio, 5.3; P < 0.001), whereas changes in right atrial pressure and pulmonary capillary wedge pressure were not. Patients with hemoconcentration had significantly lower 180-day mortality and this relationship persisted after adjustment for baseline characteristics. Thus, worsening renal function had no relationship with outcomes when occurring in patients with concomitant hemoconcentration [76]. Other studies have recently shown that higher serum creatinine levels in patients undergoing intensified diuretic treatment with either high doses of furosemide [59], rolofylline administration [70] and ultrafiltration [77].
In addition to serum creatinine, blood urea nitrogen (BUN) was shown to be an important predictor of morbidity and mortality in patients with HF [78–80]. In the randomized Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF), BUN had a stronger relationship with outcomes as compared with GFR, calculated on the basis of serum creatinine levels [81]. The major difference between serum creatinine and BUN is related to the reabsorption of BUN in the renal tubules. It is mediated by arginine vasopressin (AVP) effects on the urea transporter in the collecting duct and so it is also related to the sodium reabsorption and with volume status. Acute arterial underfilling and overdiuresis, as during intensified diuretic treatment, cause increased urea tubular reabsorption and increased BUN. BUN is also dependent on nitrogen production and in condition causing an increase in protein catabolism, such as cachexia, BUN levels are higher [82].
Thus, traditional markers of renal function, serum creatinine and BUN, are far from ideal. Markers more sensitive to renal damage and able to detect kidney damage before the development of renal dysfunction, similar to what happens with troponin measurements for the detection of myocardial damage, are needed. The results with some of the new markers of renal injury are summarized below.
Novel markers
Cystatin C
Cystatin C (CysC) is one of the novel markers of renal function. It is a 122-amino acid protein member of the family of cysteine proteinase inhibitors. It is freely filtered by the glomerulus, and reabsorbed by tubular epithelial cells where it is catabolized [83]. Thus, serum CysC is one of the best markers to estimate GFR. Multiple studies have validated the use of CysC as a renal marker in healthy adult and in patients with renal disease, and many of them found that it can more accurately estimate GFR [84–89]. Newman et al. have demonstrated that CysC is a more sensitive marker than SCr for small changes in GFR [90] and other studies further suggested that CysC is an earlier indicator of mild renal failure [87–89]. The role of CysC in patients with HF is still to be defined. In AHF, some studies have demonstrated that CysC can be a good prognostic marker. Lassus et al. measured CysC on admission and at 48 h in 292 patients hospitalized for AHF. Acute kidney injury defined by an increase in CysC >0.3 mg/l within 48 h was observed in 16% of patients and it was associated with longer length of hospitalization (P: 0.01) and with a significantly higher in-hospital mortality (odds ratio 4.0 95% CI 1.3–11.7, P: 0.01). At 90 days, the increase in CysC was an independent predictor of mortality (adjusted odds ratio 2.8 (95% CI 1.2–6.7, P: 0.02) [91].
Tubular function markers
The kidney damage in many cases not only affects the glomerulus. The tubule-interstitial function is now considered crucial and markers that are able to describe the tubule-interstitial injury have become interesting, although they have not yet used in routine clinical practice.
Neutrophil gelatinase-associated lipocalin (NGAL)
Neutrophil gelatinase-associated lipocalin (NGAL) is a small protein that is freely filtered by the glomerulus and completely reabsorbed in the tubules. It is detectable in plasma and urine, but urine concentration seems to be more accurate and not biased by bacterial sepsis or cancer [92]. Many studies have identified NGAL as an early marker of acute kidney injury [93, 94]. It has been investigated in several conditions of acute renal damage in both adult and pediatric populations, such as in critically ill patients, after cardiac surgery, in patients receiving intravenous contrast media infusion and in patients admitted to the emergency department [71, 95–102]. Also in chronic kidney disease, there is a growing literature suggesting that NGAL is associated with disease severity [103]. In patients with acute HF, NGAL can predict worsening renal function more accurately and in an earlier stage than serum creatinine. In fact, NGAL level rises about 24 h before serum creatinine values. In 91 patients admitted for AHF, worsening renal function was observed in 38% within 5 days of follow-up. Patients who developed worsening renal function had significantly higher median admission serum NGAL levels (194 ng/ml vs. 128 ng/ml, P = 0.001) and higher levels were associated with an increase in risk of developing worsening renal function [104]. Levels of urinary NGAL may be more sensitive as makers of tubular damage than serum levels [105, 106].
Kidney injury molecule 1 (KIM-1)
Kidney injury molecule-1 (KIM-1) is a marker for proximal tubular injury. It is a transmembrane protein expressed in proximal tubule cells during renal diseases associated with either proteinuria, toxic or ischemic damage. Ichimura et al. described KIM-1 in 1998 and they found that it was expressed in post-ischemic kidneys while it was undetectable in healthy subjects and showed that it is an expression of tubule-interstitial injury and inflammation [107, 108]. KIM-1 may play a role in cells regeneration process, development of interstitial fibrosis, immune response and can be involved in cell–to–cell interactions [107, 109]. Urinary KIM-1 levels correlate with tubular KIM-1 expression in experimental and in human renal disease [108, 110–112]. Also other organs express KIM-1, but to a minimal degree that would not influence renal excretion of KIM-1. In acute renal injury, KIM-1 has a strong predictive value. Liangos et al. showed that in 201 patients admitted for acute renal injury, urinary KIM-1 levels were associated with adverse clinical outcomes [113]. KIM-1 might be predictive not only in acute kidney injury but also in chronic renal disease as shown in experimental models and in patients with proteinuric chronic kidney disease [108, 111, 114]. In chronic HF, KIM-1 demonstrated a correlation with plasma N-terminal pro-brain natriuretic peptide levels and, independently of GFR values, it was associated with an increased risk of death or HF hospitalizations [106]. KIM-1 is highly sensitive to acute tubular injury but in the setting of acute HF its role is still unsettled.
N-acetyl-beta-d-glucosaminidase (NAG)
N-acetyl-beta-d-glucosaminidase (NAG) is produced in the proximal tubule and after tubular injury, it is released into the urine. It was studied in several conditions such as acute kidney injury, diabetic nephropathy, after cardiac surgery, and demonstrated a good specificity and sensitivity in detection of the tubular damage [113, 115–117]. In patients with CHF, urinary NAG levels, as KIM-1, were associated with plasma N-terminal pro-brain natriuretic peptide levels and increased risk of death or HF hospitalizations regardless of GFR, but only NAG levels were correlated with GFR (P = 0.001) and effective renal plasma flow (ERPF) (P = 0.006), suggesting that this marker can detect decreased renal perfusion in patients with low cardiac output [106]. As for KIM-1, data in AHF are lacking. The accuracy of this marker in acute kidney injury suggesting its usefulness in the acute setting and further studies are needed in order to define its role in AHF.
Conclusion
Interactions between the heart and kidney are complex and still incompletely understood. The progressive deterioration in renal function in HF patients is a result of multiple mechanisms including renal hypoperfusion caused by low cardiac output, increased renal venous and intra-abdominal pressure, neurohormonal and inflammatory activation. Several drugs have been associated with worsening renal function, namely loop diuretics, but it is still uncertain whether some agents may improve, slow or prevent, kidney damage. It may be that some of our current difficulties are related to limitations in methods of measurements of renal function. Serum creatinine levels are still universally used as marker of kidney damage or deterioration despite recent studies showing their poor sensitivity for the detection of renal damage and their limitations as prognostic variables. Novel markers of renal function may allow an earlier and more accurate detection of renal damage.
References
Thom T, Haase N, Rosamond W, et al (2006) Heart Disease and stroke Statistics—2006 Update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee [published correction appears in Circulation. 2006;113:e696]. Circulation 113:e85–e151
Haldeman GA, Croft JB, Giles WH, Rashidee A (1999) Hospitalization of patients with heart failure: National Hospital Discharge Survey, 1985 to 1995. Am Heart J 137:352–360
Cleland JG, Swedberg K, Follath F et al (2003) The EuroHeart Failure survey programme—a survey on the quality of care among patients with heart failure in Europe. Part 1: patient characteristics and diagnosis. Eur Heart J 24:442–463
Gheorghiade M, Zannad F, Sopko G, et al, for the International Working Group on Acute Heart Failure Syndromes (2005) Acute heart failure syndromes: current state and framework for future research. Circulation 112:3958–3968
Gheorghiade M, Gattis WA, O’Connor CM, et al, for the Acute and Chronic Therapeutic Impact of a Vasopression Antagonist in Congestive Heart Failure (ACTIV in CHF) Investigators (2004) Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: a randomized controlled trial. JAMA 291:1963–1971
Adams KF Jr, Fonarow GC, Emerman CL, et al, for the ADHERE Scientific Advisory Committee and Investigators (2005) Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J 149:209–216
Hillege HL, Girbes AR, de Kam PJ et al (2000) Renal function, neurohormonal activation, and survival in patients with chronic heart failure. Circulation 102:203–210
Dries DL, Exner DV, Domanski MJ et al (2000) The prognostic implications of renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol 35:681–689
Jones CA, McQuillan GM, Kusek JW, Eberhardt MS, Herman WH, Coresh J, Salive M, Jones CP, Agodoa LY (1998) Serum creatinine levels in the US population: third National Health and Nutrition Examination Survey. Am J Kidney Dis 32:992–999
Nohria A, Hasselblad V, Stebbins A et al (2008) Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol 51:1268–1274
Ahmed A, Rich MW, Sanders PW, Perry GJ, Bakris GL, Zile MR et al (2007) Chronic kidney disease associated mortality in diastolic versus systolic heart failure: a propensity matched study. Am J Cardiol 99:393–398
Hillege HL, Nitsch D (2006) Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation 113:671–678
Zannad F, McMurray JJ, Krum H et al (2011) Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 6(364):11–21
Butler J, Chirovsky D, Phatak H et al (2010) Renal function, health outcomes, and resource utilization in acute heart failure: a systematic review. Circ Heart Fail 3:726–745
Smith GL, Lichtman JH, Bracken MB et al (2006) Renal impairment and outcomes in heart failure: systematic review and meta-analysis. J Am Coll Cardiol 47:1987–1996
Gottlieb SS, Abraham W, Butler J et al (2002) The prognostic importance of different definitions of worsening renal function in congestive heart failure. J Card Fail 8:136–141
Forman DE, Butler J, Wang Y et al (2004) Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J Am Coll Cardiol 43:61–67
Damman K, Navis G, Voors AA, Asselbergs FW, Smilde TD, Cleland JG, van Veldhuisen DJ, Hillege HL (2007) Worsening renal function and prognosis in heart failure: systematic review and meta-analysis. J Card Fail 13:599–608
Griendling KK, Minieri CA, Ollerensaw JD, Alexander RW (1994) Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74:1141–1148
Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM (2003) Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 41:2164–2171
Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK (2003) Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int 63:179–185
Masuo K, Lambert GW, Esler MD, Rakugi H, Ogihara T, Schlaich MP (2010) The role of nervous activity in renal injury and end-stage renal disease. Hypertens Res 33(6):521–528
Krum H, Schlaich M, Whitbourn R et al (2009) Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet 373:1275–1281
McClellen W, Aronoff SL, Bolton WK et al (2004) The prevalence of anemia in patients with chronic kidney disease. Curr Med Res Opin 20:1501–1510
Young JB, Abraham WT, Albert NM, Gattis Stough W, Gheorghiade M, Greenberg BH, O’Connor CM, She L, Sun JL, Yancy CW, Fonarow GC, for the OPTIMIZE-HF Investigators and Coordinators (2008) Relation of low hemoglobin and anemia to morbidity and mortality in patients hospitalized with heart failure (insight from the OPTIMIZE-HF registry). Am J Cardiol 101:223–230
George J, Patal S, Wexler D et al (2005) Circulating erythropoietin levels and prognosis in patients with congestive heart failure: comparison with neurohormonal and inflammatory markers. Arch Intern Med 165:1304–1309
Calvillo L, Latini R, Kajstura J et al (2003) Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sci USA 100:4802–4806
Vesey DA, Cheung C, Pat B et al (2004) Erythropoietin protects against ischaemic acute renal injury. Nephrol Dial Transplant 19:348–355
Damman K, Navis G, Smilde TD, Voors AA, van der Bij W, van Veldhuisen DJ, Hillege HL (2007) Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail 9(9):872–878
McCullough PA, Ahmad A (2001) Cardiorenal syndromes. World J Cardiol 26;3(1):1–9
Mullens W, Abrahams Z, Francis GS et al (2009) Importance of venous congestion for worsening renal function in advanced decompensated heart failure. J Am Coll Cardiol 53:589–596
Damman K, van Deursen VM, Navis G et al (2009) Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol 53:582–588
Mullens W, Abrahams Z, Skouri HN et al (2008) Elevated intra-abdominal pressure in acute decompensated heart failure. J Am Coll Cardiol 51:300–306
Damman K, Voors AA, Hillege HL et al (2010) Congestion in chronic systolic heart failure is related to renal dysfunction and increased mortality. Eur J Heart Fail 12:974–982
Uthoff H, Breidthardt T, Klima T et al (2011) Central venous pressure and impaired renal function in patients with acute heart failure. Eur J Heart Fail 13(4):432–439
Funaya H, Kitakaze M, Node K, Minamino T, Komamura K, Hori M (1997) Plasma adenosine levels increase in patients with chronic heart failure. Circulation 95:1363–1365
Metra M, Brutsaert D, Dei Cas L, Gheorghiade M (2011) Chapter 49 acute heart failure: epidemiology, classification, and pathophysiology. The ESC textbook of acute and intensive cardiac care 1(1):med-9780199584314-chapter–med-9780199584314-chapter
Metra M, O’Connor CM, Davison BA, et al (2011) Early dyspnoea relief in acute heart failure: prevalence, association with mortality, and effect of rolofylline in the PROTECT Study. Eur Heart J, 8 Mar 2011 [Epub ahead of print]
Knight EL, Glynn RJ, McIntyre KM et al (1999) Predictors of decreased renal function in patients with heart failure during angiotensin-converting enzyme inhibitor therapy: results from the studies of left ventricular dysfunction (SOLVD). Am Heart J 138:849–855
Metra M, Nodari S, Parrinello G et al (2008) Worsening renal function in patients hospitalised for acute heart failure: clinical implications and prognostic significance. Eur J Heart Fail 10:188–195
De Silva R, Nikitin NP, Witte KKA et al (2006) Incidence of renal dysfunction over 6 months in patients with chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J 27:569–581
Mielniczuk LM, Tsang SW, Desai AS et al (2008) The association between high-dose diuretics and clinical stability in ambulatory chronic heart failure patients. J Card Fail 14:388–393
Felker GM, O’Connor CM, Braunwald E; Heart Failure Clinical Research Network Investigators (2009) Loop diuretics in acute decompensated heart failure: necessary? Evil? A necessary evil? Circ Heart Fail 2:56–62
Metra M, Bugatti S, Bettari L, et al (2011) Can we improve the treatment of congestion in heart failure? Expert Opin Pharmacother, 23 Feb 2011 [Epub ahead of print]
Jentzer JC, DeWald TA, Hernandez AF (2010) Combination of loop diuretics with thiazide-type diuretics in heart failure. J Am Coll Cardiol 56:1527–1534
Hasselblad V, Gattis Stough W, Shah MR et al (2007) Relation between dose of loop diuretics and outcomes in a heart failure population: results of the ESCAPE trial. Eur J Heart Fail 9:1064–1069
Felker et al (2011) Determining optimal dose and duration of diuretic treatment in people with acute heart failure (DOSE-AHF). N Engl J Med (in press)
Maskin CS, Ocken S, Chadwick B, LeJemtel TH (1985) Comparative systemic and renal effects of dopamine and angiotensin-converting enzyme inhibition with enalaprilat in patients with heart failure. Circulation 72:846–852
Elkayam U, Ng TM, Hatamizadeh P et al (2008) Renal vasodilatory action of dopamine in patients with heart failure: magnitude of effect and site of action. Circulation 117:200–205
Friedrich JO, Adhikari N, Herridge MS, Beyene J (2005) Meta-analysis: low-dose dopamine increases urine output but does not prevent renal dysfunction or death. Ann Intern Med 142:510–524
Kellum JA, Decker J (2001) Use of dopamine in acute renal failure: a meta-analysis. Crit Care Med 29:1526–1531
Giamouzis G, Butler J, Starling RC et al (2010) Impact of dopamine infusion on renal function in hospitalized heart failure patients: results of the Dopamine in Acute Decompensated Heart Failure (DAD-HF) Trial. J Card Fail 16:922–930
Leier CV, Heban PT, Huss P et al (1978) Comparative systemic and regional hemodynamic effects of dopamine and dobutamine in patients with cardiomyopathic heart failure. Circulation 58:466–475
Mitrovic V, Hernandez AF, Meyer M, Gheorghiade M (2009) Role of guanylate cyclase modulators in decompensated heart failure. Heart Fail Rev 14:309–319
Publication Committee for the VMAC Investigators (Vasodilatation in the Management oacknerf Acute CHF) (2002) Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 287:1531–1540
O’Connor CM, Hernandez AF, Starling RC et al (2009) Rationale and design of the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure Trial (ASCEND-HF). Am Heart J 157:271–277
Hunt SA, Abraham WT, Chin MH et al (2009) Focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 119:e391–e479
Lindenfeld J, Albert NM, Boehmer JP et al (2010) Executive summary: HFSA 2010 comprehensive heart failure practice guideline. J Card Fail 16:475–539
Voors AA, van Veldhuisen DJ (2010) Cardiorenal effects of recombinant human natriuretic peptides. J Am Coll Cardiol 55:1852–1853
Wang DJ, Dowling TC, Meadows D et al (2004) Nesiritide does not improve renal function in patients with chronic heart failure and worsening serum creatinine. Circulation 110:1620–1625
Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K (2005) Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA 293:1900–1905
Hernandez AF, O’Connor CM, Starling RC et al (2011) Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure Trial (ASCEND-HF). New Engl J Med (in press)
Suwa M, Seino Y, Nomachi Y, Matsuki S, Funahashi K (2005) Multicenter prospective investigation on efficacy and safety of carperitide for acute heart failure in the ‘real world’ of therapy. Circ J 69(3):283–290
Ren Y, Garvin JL, Carretero OA (2001) Efferent arteriole tubuloglomerular feedback in the renal nephron. Kidney Int 59:222–229
Givertz MM, Massie BM, Fields TK, Pearson LL, Dittrich HC; CKI-201 and CKI-202 Investigators (2007) The effects of KW-3902, an adenosine A1-receptor antagonist, on diuresis and renal function in patients with acute decompensated heart failure and renal impairment or diuretic resistance. J Am Coll Cardiol 50:1551–1560
Cotter G, Dittrich HC, Weatherley BD et al (2008) The PROTECT pilot study: a randomized, placebo-controlled, dose-finding study of the adenosine A1 receptorantagonist rolofylline in patients with acute heart failure and renal impairment. J Card Fail 14:631–640
Metra M, O’Connor CM, Davison BA, et al (2011) Early dyspnoea relief in acute heart failure: prevalence, association with mortality, and effect of rolofylline in the PROTECT Study. Eur Heart J, ehr042 first published online 8 Mar 2011. doi:10.1093/eurheartj/ehr042
Massie BM, O’Connor CM, Metra M et al (2010) Rolofylline, an adenosine A1—receptor antagonist, in acute heart failure. N Engl J Med 363:1419–1428
Bonventre JV, Vaidya VS, Schmouder R, Feig P, Dieterle F (2010) Next-generation biomarkers for detecting kidney toxicity. Nat Biotechnol 28:436–440
Herget-Rosenthal S, Marggraf G, Hüsing J et al (2004) Early detection of acute renal failure by serum cystatin C. Kidney Int 66:1115–1122
Haase-Fielitz A, Bellomo R, Devarajan P et al (2009) Novel and conventional serum biomarkers predicting acute kidney injury in adult cardiac surgery—a prospective cohort study. Crit Care Med 37:553–560
Waikar SS, Bonventre JV (2008) Biomarkers for the diagnosis of acute kidney injury. Nephron Clin Pract 109(4):c192–c197
Stevens LA, Coresh J, Greene T, Levey AS (2006) Assessing kidney function-measured and estimated glomerular filtration rate. N Engl J Med 354:2473–2483
Kunzendorf U, Haase M, Rölver L, Haase-Fielitz A (2010) Novel aspects of pharmacological therapies for acute renal failure. Drugs 70:1099–1114
Waikar SS, Bonventre JV (2009) Creatinine kinetics and the definition of acute kidney injury. J Am Soc Nephrol 20:672–679
Testani JM, Chen J, McCauley BD, Kimmel SE, Shannon RP (2010) Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation 122:265–272
Costanzo MR, Guglin ME, Saltzberg MT et al (2007) Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J Am Coll Cardiol 49:675–683
Filippatos G, Rossi J, Lloyd-Jones DM et al (2007) Prognostic value of blood urea nitrogen in patients hospitalized with worsening heart failure: insights from the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in CHF) study. J Card Fail 13:360–364
Cauthen CA, Lipinski MJ, Abbate A et al (2008) Relation of blood urea nitrogen to longterm mortality in patients with heart failure. Am J Cardiol 101:1643–1647
Aronson D, Mittleman MA, Burger AJ (2004) Elevated blood urea nitrogen level as a predictor of mortality in patients admitted for decompensated heart failure. Am J Med 116:466–473
Klein L, Massie BM, Leimberger JD et al (2008) Admission or changes in renal function during hospitalization for worsening heart failure predict postdischarge survival: results from the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF). Circ Heart Fail 1:25–33
Schrier RW (2008) Blood urea nitrogen and serum creatinine: not married in heart failure. Circ Heart Fail 1:2–5
Grubb A (1992) Diagnostic value of analysis of cystatin C and protein HC in biological fluids. Clin Nephrol 38:S20–S27
Newman DJ, Thakkar H, Edwards RG, Wilkie M, White T, Grubb A et al (1995) Serum cystatin C measured by automated immunoassay: a more sensitive marker of changes in GFR than serum creatinine. Kidney Int 47:312–318
Kyhse-Andersen J, Schmidt C, Nordin G, Andersson B, Nilsson Ehle P, Lindstrom V et al (1994) Serum cystatin C, determined by a rapid, automated particle-enhanced turbidimetric method, is a better marker than serum creatinine for glomerular filtration rate. Clin Chem 40:1921–1926
Grubb A, Simonsen O, Sturfelt G et al (1985) Serum concentration of cystatin C, factor D and 2-microglobulin as a measure of glomerular filtration rate. Acta Med Scand 218:499–503
Randers E, Erlandsen EJ, Pedersen OL, Hasling C, Danielsen H (2000) Serum cystatin C as an endogenous parameter of the renal function in patients with normal to moderately impaired kidney function. Clin Nephrol 54:203–209
Meir P, Froidevaux C, Dayer E, Blanc E (2001) Cystatin C concentration and glomerular filtration rate. Lancet 357:634–635
Herget-Rosenthal S, Trabold S, Pietruck F, Heemann U, Philipp T, Kribben A (2000) Cystatin C: efficacy as screening test for reduced glomerular filtration rate. Am J Nephrol 20:97–102
Newman DJ, Thakkar H, Edwards RG, Wilkie M, White T, Grubb AO et al (1994) Serum cystatin C: a replacement for creatinine as a biochemical marker for GFR. Kidney Int 46:S20–S21
Lassus JP, Nieminen MS, Peuhkurinen K et al (2010) Markers of renal function and acute kidney injury in acute heart failure: definitions and impact on outcomes of the cardiorenal syndrome. Eur Heart J 31:2791–2798
Kai M, Schmidt-Ott KM, Mori K, Li JY et al (2007) Dual action of neutrophil gelatinase—associated lipocalin. J Am Soc Nephrol 18:407–413
Mishra J, Ma Q, Prada A et al (2003) Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 14:2534–2543
Supavekin S, Zhang W, Kucherlapati R et al (2003) Differential gene expression following early renal ischemia/reperfusion. Kidney Int 63:1714–1724
Zappitelli M, Washburn KK, Arikan AA, et al (2007) Urine neutrophil gelatinase-associated lipocalin is an early marker of acute kidney injury in critically ill children: a prospective cohort study. Crit Care 11:R84
Wheeler DS, Devarajan P, Ma Q et al (2008) Serum neutrophil gelatinase-associated lipocalin (NGAL) as a marker of acute kidney injury in critically ill children with septic shock. Crit Care Med 36:1297–1303
Wagener G, Jan M, Kim M et al (2006) Association between increases in urinary neutrophil gelatinase-associated lipocalin and acute renal dysfunction after adult cardiac surgery. Anesthesiology 105:485–491
Bennett M, Dent CL, Ma Q et al (2008) Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clin J Am Soc Nephrol 3:665–673
Mishra J, Dent C, Tarabishi R et al (2005) Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 365:1231–1238
Ling W, Zhaohui N, Ben H et al (2008) Urinary IL-18 and NGAL as early predictive biomarkers in contrast-induced nephropathy after coronary angiography. Nephron Clin Pract 108:c176–c181
Bachorzewska-Gajewska H, Malyszko J, Sitniewska E, Malyszko JS, Dobrzycki S (2006) Neutrophil-gelatinase-associated lipocalin and renal function after percutaneous coronary interventions. Am J Nephrol 26:287–292
Nickolas TL, O’Rourke MJ, Yang J et al (2008) Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med 148:810–819
Bolignano D, Coppolino G, Campo S, Aloisi C, Nicocia G, Frisina N et al (2008) Urinary neutrophil gelatinase-associated lipocalin (NGAL) is associated with severity of renal disease in proteinuric patients. Nephrol Dial Transpl 23:414–416
Aghel A, Shrestha K, Mullens W, Borowski A, Tang WH (2010) Serum neutrophil gelatinase-associated lipocalin (NGAL) in predicting worsening renal function in acute decompensated heart failure. J Card Fail 16:49–54
Damman K, van Veldhuisen DJ, Navis G, Voors AA, Hillege HL (2008) Urinary neutrophil gelatinase associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failure. Eur J Heart Fail 10:997–1000
Damman K, van Veldhuisen DJ, Navis G et al (2010) Tubular damage in chronic systolic heart failure is associated with reduced survival independent of glomerular filtration rate. Heart 96:1297–1302
Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL et al (1998) Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 27:4135–4142
Van Timmeren MM, Van den Heuvel MC, Bailly V, Bakker SJ, Van GH, Stegeman CA (2007) Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J Pathol 212:209–217
Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, Umetsu SE et al (2005) TIM-4 is the ligand for TIM-1, and the TIM-1–TIM-4 interaction regulates T cell proliferation. Nat Immunol 6:455–464
Kramer AB, van Timmeren MM, Schuurs TA, Vaidya VS, Bonventre JV, van Goor H et al (2009) Reduction of proteinuria in adriamycin-induced nephropathy is associated with reduction of renal kidney injury molecule-1 (Kim-1) over time. Am J Physiol Renal Physiol 276:1136–1145
Van Timmeren MM, Bakker SJ, Vaidya VS, Bailly V, Schuurs TA, Damman J et al (2006) Tubular kidney injury molecule-1 in protein-overload nephropathy. Am J Physiol Renal Physiol 291:456–464
Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, Bonventre JV (2006) Urinary kidney injury molecule-1: a sensitive quantitative biomarker for early detection of kidney tubular injury. Am J Physiol Renal Physiol 290:517–529
Liangos O, Perianayagam MC, Vaidya VS, Han WK, Wald R, Tighiouart H et al (2007) Urinary N-acetyl-β-d-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J Am Soc Nephrol 18:904–912
Waanders F, Vaidya VS, van Goor H, Leuvenink H, Damman K, Hamming I et al (2009) Effect of renin-angiotensin-aldosterone system inhibition, dietary sodium restriction, and/or diuretics on urinary kidney injury molecule 1 excretion in nondiabetic proteinuric kidney disease: a post hoc analysis of a randomized controlled trial. Am J Kidney Dis 53:16–25
Parikh CR, Lu JC, Coca SG, Devarajan P (2010) Tubular proteinuria in acute kidney injury: a critical evaluation of current status and future promise. Ann Clin Biochem 47:301–312
Nauta FL, Boertien WE, Bakker SJ et al (2011) Glomerular and tubular damage markers are elevated in patients with diabetes. Diabetes Care 34(4):975–981
Hamada Y, Kanda T, Anzai T et al (1999) N-acetyl-beta-d-glucosaminidase is not a predictor, but an indicator of kidney injury in patients with cardiac surgery. J Med 30:329–336
Conflict of interest
Prof M. Metra received honoraria for speeches and advisory board meetings from Corthera, Novartis and Servier. Drs. V. Carubelli, C. Lombardi, L. Bettari, S. Bugatti, V. Lazzarini and Prof L. Dei Cas have no conflicts of interest or financial ties to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Carubelli, V., Metra, M., Lombardi, C. et al. Renal dysfunction in acute heart failure: epidemiology, mechanisms and assessment. Heart Fail Rev 17, 271–282 (2012). https://doi.org/10.1007/s10741-011-9265-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-011-9265-z