
Abstract
End-stage heart failure is characterized by a number of abnormalities at the cellular level, which include changes in excitation–contraction coupling, alterations in contractile proteins and activation/deactivation of signaling pathways. Even though many of these changes are adaptive to the high workload and stress in heart failure, a significant number of these alterations are deeply deleterious to the cardiac cell. In this article, we will review the changes in calcium cycling that occur in myopathic hearts and how they can be effectively targeted. We will also focus on protein misfolding in the setting of cardiac dysfunction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure is characterized by deficiencies in contraction of the affected ventricles and the single cardiac myocytes [1, 2]. Force and shortening of cardiomyocytes are tightly regulated by the mechanisms of excitation–contraction (E–C) coupling [3]. A number of abnormalities have been described at various steps of E–C coupling of cardiomyocytes isolated from failing hearts as well as Ca2+ disregulation [4, 5].
Ca2+ concentrations within the cellular compartments is used across cell types from prokariotes to eukariotes as signaling molecule that control virtually every cell function from motility to cell death. Ca2+ compartimentalization within cellular domains and tightly regulated dynamic changes regulate Ca2+ dependent cellular functions and its disruption is more and more recognized at the basis of devastating cellular outcomes.
Ca2+ cycling has been found to be critically dysregulated in failing hearts with increased [Ca2+]i and decreased sarcoplasmic reticulum (SR) Ca2+ content ([Ca2+]ER/SR) leading to a decrease in systolic function and abnormal diastolic function [3, 6, 7]. Unloaded [Ca2+]SR in turn regulates the effectors of SR Ca2+ release further worsening the abnormal Ca2+ distribution. In this article, we will summarize the abnormalities in E–C coupling in failing hearts and then focus on the disregulation of the SR Ca2+ homeostasis induced endoplasmic reticulum (ER) stress response.
ER function in control of contractility
Excitation–contraction coupling
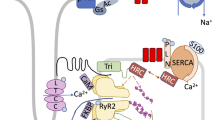
In the mammalian heart, depolarization of the cell membrane leads to the opening of voltage gated L-type Ca2+ channels, located in the T-tubular regions of the myocytes, allowing the influx of Ca2+ into the cell [3, 7] (Fig. 1). These channels are in close proximity to the Ca2+ release channels, known as ryanodine receptors (RyRs), which are located on the SR, the major Ca2+ storing organelle in the cardiomyocyte. Ca2+ entering the cells through a single L-type Ca2+ channel induces the opening of one or a cluster of RyRs resulting in the local release of Ca2+ from the SR [3, 7–9]. During membrane depolarization, a large number of L-type Ca2+ channels are opened, resulting in a large release of Ca2+ from the RyRs, raising cytosolic Ca2+ from 0.1–0.2 mM to 2–10 mM.

Excitation–contraction coupling in mammalian myocardial cell
The coupling between the L-type Ca channel and the RyRs has been modeled and experimentally characterized as a functional Ca2+ signaling and releasing unit, namely the Ca2+ spark. Ca2+ sparks were first identified as the “elementary events” of spontaneous increases in intracellular [Ca2+], which were detected by laser scanning confocal microscopy sensitive fluorescent Ca2+ indicators. Functionally, the Ca2+ sparks represent Ca2+ releases from the SR through the opening of the SR Ca2+ release channels/RyRs. Ca2+ sparks are produced by 10–100 RyRs based on the ratio of the Ca2+ sparks current and the single RyR channel current and because the morphology of Ca2+ sparks vary even within a single cell. Ca2+ sparks are depicted/measured by their morphology and the frequency of occurrence. The information of the morphology includes the sizes of Ca2+ sparks (amplitude, width, and duration) and the kinetics is described by the spark rising and decaying dynamics. The activity of individual RyRs and the number of the RyRs recruited during a spark play an important role in the spark morphology. Thus, the more active the Ca2+ release channels are, the more Ca2+ would be released and as a consequent, the increased size and the more occurrence of the Ca2+ sparks would be observed. Therefore, direct modifications that change the RyRs activity or the modulators that regulate RyR activity will have an impact on the size and frequency of Ca2+ sparks. SR Ca2+ content also regulates Ca2+ sparks because luminal [Ca2+] plays a critical role in regulating RyR by increasing the activity of the RyRs and by sensitizing the threshold of RyR for activation to the stimulus. Ca2+ sparks can be triggered by the Ca2+ influx through the L-type Ca2+ sparks during voltage pulses. Thus, the kinetics, fidelity, and stoichiometry of coupling between L-type Ca2+ channels and RyR plays a critical role in determination of the signal transduction during E–C coupling process. The distance of the cleft of between the cell plasma membrane and the SR membrane, which is roughly 12 nm in normal cardiomyocytes, is very critical for such a kind of coupling and signal transduction. Extension of the distance between them by pulling the plasma membrane under tight-seal condition decreases/abolishes the signaling reliability.
The release of Ca2+ into the myofibrillar space in turn results in cross-bridge formation and contraction [10–13]. The myofilaments are organized in a regular array of thick and thin filaments giving the typical striated appearance of the entire myocyte. Each unit of this striated organization, known as sarcomere, is composed of one unit of interacting thin and thick filaments. The thick filaments consist mainly of myosin heavy chains and two pairs of light chains. The “tail” of the molecule is coiled with two myosin heavy chains wound around each other. The “heads” of myosin include the globular region of one myosin molecule and two myosin light chains [10–13]. Each myosin head has an ATP-binding area in close proximity to myosin ATPase, which breaks down ATP. Two myosin heavy chain isoforms exist in human mammalian myocardium, α- and β-MHC. In human ventricular myocardium, only 10–20% of α-MHC while the slower β-MHC is more abundant. The thin filaments are comprised of actin and troponin complex. Within the sarcomere, the actin polymers intertwine in a helical fashion. At intervals of 385 Å along the thin filaments are a group of three regulatory proteins called troponin complex carried on a long helical molecule called tropomyosin. The troponin complexes are made up of one molecule each of troponin C, troponin I, and troponin T. The strength of the bond linking troponin I and actin varies depending on the intracellular Ca2+ level.
When the cytosolic Ca2+ is low the tropomyosin–troponin complex is positioned in such a way that the myosin heads cannot interact with actin. This is due to the bond linking troponin I to actin. When cytosolic Ca2+ increases, it binds to troponin C strengthening the bond between troponin C and troponin I and weakening the bond linking troponin I to actin. This leads to a conformational change of the tropomyosin–troponin C complex, which allows the myosin head to interact with actin. When Ca2+ binds to troponin C, it exposes the active site on the thin filament, permitting the myosin head to bind weakly to the actin filaments. Subsequent ATP hydrolysis allows a strong binding of the myosin head to actin, which is followed by a power stroke that moves the actin molecule, 5–10 nm, locking the myosin head into a rigor state. The myosin head releases ADP and is ready to accept ATP, which starts the cycle again. ATP binding to the myosin head dissociates the thin and thick filaments.
Relaxation occurs when Ca2+ detaches from troponin C and is re-accumulated back into the SR by the cardiac isoform of the Ca2+-ATPase pump (SERCA2a) and extruded extracellularly by the sarcolemmal Na+/Ca2+ exchanger. The contribution of each of these mechanisms for lowering cytosolic Ca2+ varies among species. In humans ∼75% of the Ca2+ is removed by SERCA2a and ∼25% by the Na+/Ca2+ exchanger. SERCA2a transports Ca2+ back to the luminal space of the SR against a Ca2+ gradient by an energy dependent mechanism (one molecule of ATP is hydrolyzed for the transport of two molecules of Ca2+), where it binds to a Ca2+ buffering protein, calsequestrin. The Ca2+ pumping activity of SERCA2a is regulated by phospholamban. In its unphosphorylated state, phospholamban inhibits the Ca2+-ATPase, whereas phosphorylation of phospholamban by cAMP-dependent protein kinase and by Ca2+-calmodulin dependent protein kinase reverses this inhibition.
Ca2+ is removed from the cytosol by the sarcolemmal Na+/Ca2+ exchanger, which has high capacity, but low affinity and is the major Ca2+ extrusion mechanism of the cardiac myocyte [3]. This system returns Ca2+ concentrations to diastolic levels (∼100–300 nM) and may, therefore, contribute significantly to myocardial relaxation.
Ca2+ handling in failing hearts
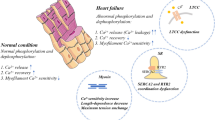
The myopathic heart exhibits abnormalities in both the systolic and diastolic phase (Fig. 2). Changes in diastolic function often appear earlier than systolic dysfunction. In fact, compensated hypertrophy phenotypically demonstrates impaired relaxation parameters in the presence of normal or increased systolic function [1]. Abnormalities in Ca2+ handling were noted more than 15 years ago when Ca2+ transients recorded with the Ca2+ indicator aequorin from trabeculae from myopathic human hearts removed at the time of cardiac transplantation revealed significantly prolonged Ca2+ transient with an elevated end-diastolic intracellular Ca2+ [1]. These defects were subsequently found in single isolated cardiomyocytes loaded with the fluorescent indicator Fura-2 from myopathic hearts [14]. The Ca2+ transients were characterized as having elevated diastolic Ca2+ levels, a decreased systolic Ca2+, and prolonged relaxation phase. Studies both in muscle strips and isolated cardiomyocytes found that systolic Ca2+ concentration were decreased in the failing state while diastolic Ca2+ concentration were elevated [14]. These differences were accentuated at higher stimulation rates.

Abnormalities in EC coupling mechanisms in the setting of heart failure. The figures depicts the various changes in ion channels in the setting of heart failure
The slow relaxation and abnormal force–frequency relationship observed in isolated muscles as well as in isolated myocytes from failing hearts, suggest a deficiency in Ca2+ reuptake by the SR. Ca2+ transport into the SR occurs via the SR Ca2+ ATPase Ca2+ pump (SERCA2a). Failing hearts have been characterized by defects in SR function. Specifically, “relaxation abnormalities” correlated with deficient SR Ca2+ uptake have been associated with a decreased expression level of SR Ca2+-ATPase and reduction in SR Ca2+-ATPase activity [15–19]. A number of investigators have shown that the levels of SERCA2a message and protein to be consistently decreased in heart failure [16, 19–22] in relation to phospholamban. Associated with a decrease in mRNA, there is a decrease in SR Ca2+-ATPase activity and SR Ca2+ uptake from SR vesicles and membranes isolated from failing human heart. Indeed, in experiments where the SR vesicles were isolated from human hearts, vesicles from failing human hearts had decreased rates of Ca2+ uptake when compared to normal hearts [15, 18, 23–25]. Furthermore, the SR Ca2+ ATPase activity is inversely related to diastolic Ca2+ [16, 19, 20, 26–29]. In failing hearts diastolic Ca2+ is elevated while ATPase activity is decreased. In failing hearts from human and experimental models of heart failure, phosphorylation of phospholamban at both serine 16 and threonine 17 are decreased. This results in further inhibition of the remaining functional SERCA2a pump in the failing heart.
Two reports have revealed an association between PL mutations and cardiomyopathy [30, 31]. The inheritance of the PL mutation encoding Arg9Cys was linked to the dominant inheritance of dilated cardiomyopathy in a large American family [31]. The effects of the PL Arg9Cys mutation were characterized by expression in heterologous cell culture, by the creation of a transgenic mouse [31] and by analysis of cardiac tissue obtained from an explanted heart [31]. In all cases, the level of PL phosphorylation was reduced markedly. The key effect of the mutation was enhancement of the affinity of Arg9Cys mutant PL for PKA. In attempting to phosphorylate mutant PL, PKA becomes trapped in a stabilized mutant PL–PKA complex and can no longer dissociate to phosphorylate wild-type PL molecules [31]. The effect seems to be local and restricted to the SR, perhaps because a specific fraction of PKA is associated with the SR through A-kinase anchoring proteins. These results explain the dominant effects of the mutation. Affected individuals must go through life with chronically inhibited SERCA2a and can never draw on their full cardiac reserve. A second human PL mutation, Leu39stop, was discovered in two large Greek families [30]. The heterozygous inheritance of the Leu39stop mutation in one family led to left ventricular hypertrophy in one-third of the older affected family members, without diminished contractile performance. However, the inheritance of two copies of the mutant PL gene led to dilated cardiomyopathy and heart failure in two teenage siblings. In heterologous expression studies, the Leu39stop mutant protein was unstable or misrouted to other membranes and no protein was detected in the SR of these cells or in the explanted heart from one of the affected individuals. As a result, there was no effect of the mutant protein, in either the homozygous or heterozygous state, on the Ca2+ affinity of SERCA2a. Accordingly, these two homozygous mutant individuals can be considered to be equivalent to a PL-null genotype with a phenotype of dilated cardiomyopathy. So, in contrast to the benefits of PL ablation in mouse, humans that lack PL develop lethal cardiomyopathy. A caveat in these studies is that the number of affected individuals is very low and the lod score for linkage of the mutation to the disease is low.
As mentioned above, there is a decrease in PLN phosphorylation in heart failure. An increase in SR protein phosphatase 1 activity in patients with end-stage heart failure, which coupled with the downregulation of β-receptors, contributes directly to the decreased PLN phosphorylation and increased inhibition of SERCA2a’s Ca2+ affinity. Studies in experimental and human heart failure indicate that these increases in phosphatase activity are accompanied by increases in the dephosphorylated state of its endogenous inhibitor I-1. The dephosphorylated I-1 is inactive, but upon phosphorylation by PKA on Thr35, it selectively and potently inhibits PP1 catalytic activity with IC50 of 1 nM. Thus, regulation of the phosphatase activity in SR may provide another mechanism by which the cell achieves increases in PLN phosphorylation and thereby, increases in basal contractile parameters and their responses to β-agonists. Actually, during β-adrenergic stimulation, the increases in cAMP-dependent protein kinase activity result in increased phosphorylation of the I-1 protein at T35, which then effectively inhibits the SR phosphatase activity, allowing for amplification of the β-agonist cascade stimulatory effects through “unopposed” PLN phosphorylation [32, 33]. However, in heart failure, the I-1 is dephosphorylated due to decreased β-adrenergic drive and this result in diminished inhibition of the type 1 phosphatase and dephosphorylation of PLN [34]. Ablation of I-1 results in increased protein phosphatase activity, which is associated with decreases in PLN phosphorylation status and thus, increased inhibition of SERCA2a’s Ca2+ affinity and myocardial contractility. Overexpression of the constitutively active I-1 had opposite effects than its ablation [35]. Intriguingly, the Thr35 residue of I-1 is dephosphorylated by PP2B (calcineurin) in the hippocampus and this represents a unique scenario of fine-tuning the regulation of PP1 activity, via its I-1 protein, integrating a cross talk between PKA and calcineurin.
The conductive properties of both the L-type Ca2+ channels and the ryanodine release channels from patients with dilated cardiomyopathy, as assayed by voltage clamp are essentially normal, but their coupling may be defective, such that in failing myocardial cells L-type Ca2+ channels have a reduced ability to activate the adjacent RyRs. Changes of Ca2+ spark characteristics have been investigated in explanted failing human hearts as well as different animal models of cardiac hypertrophy and heart failure. Overall, a number of findings have been noted across different models of heart failure: (1) the efficiency of the signaling transduction between L-type Ca2+ channel and RyR was reduced [36]. This finding has been interpreted as a defective communication between L-type Ca2+ channels and RyRs, either because of the mismatch of the two Ca2+ channels or an increase in the distance between LCC and RyR has been changed. (2) [Ca2+]i transient is reduced secondary to a decrease in the SR Ca2+ load. (3) The frequency of Ca2+ sparks occurrence is reduced in failing myocytes while the triggering L-type Ca2+ current remains unchanged. (4) The rising and decaying time is also elongated in failing myocytes. All these changes in Ca2+ sparks suggest that several molecular mechanisms, including increased RyR activity (increased duration and width of Ca2+ sparks), reduction in SR Ca2+ content (reduced amplitude of Ca2+ sparks and the frequency of Ca2+ sparks responded to field stimulation), as well as the defective E–C coupling gain (reduced frequency of Ca2+ sparks responded to field stimulation). In heart failure, the stimulation of the sympathetic nervous system results in phosphorylation of RyR2 by PKA. PKA phosphorylation modulates RyR2 function by changing the sensitivity of RyR2 to Ca2+ resulting in “leaky” channels, which may cause diastolic Ca2+ release, and generating delayed after depolarizations. In addition, PKA hyperphosphorylation of RyR2s in failing functionally uncouples the channels from the L-type Ca2+ channel thereby reducing EC coupling gain.
Most studies indicate that mRNA and protein levels of the Na+/Ca2+ exchanger are increased in human myopathic hearts, but not in all preparations [3, 37–39]. Likewise, studies find the functional capacity of the Na+/Ca2+ exchanger to transport Ca2+ is increased. The consequence of an increased activity of the Na+/Ca2+ exchanger in failing hearts may be to compensate for the reduction in SR Ca2+-ATPase activity. Increased activity of the Na+/Ca2+ exchanger should aid in myocardial relaxation, albeit at the cost of reduced Ca2+ release from the SR during systole. This would be particularly evident at higher rates of stimulation, and thus lead to a blunted frequency response as is commonly seen in myopathic human myocardium.
The Na+/Ca2+ exchanger can operate to bring Ca2+ into the cell or extrude Ca2+ out from the cell [3, 26, 37–39]. There is an increase in sensitivity to compounds that produce positive inotropic effects through raised intracellular Na+, either by inhibiting the Na/K-ATPase or by opening Na+ channels, in muscle strips from failing human hearts. It has been suggested that the relaxation abnormalities produced by the loss of SERCA2a activity could be, at least partially, compensated for by an increase in the activity of the Na+/Ca2+ exchanger.
As shown in Fig. 3, the multiple abnormalities that are found in failing cardiomyocytes have been targeted by various manipulations. These old and novel strategies have been designed to enhance contractile function in the heart.

Various targets within the cardiomyocytes that can be affected to improve cardiac function
ER function in control of protein synthesis and post-translational modification
In addition to his central role in controlling excitation–contraction coupling in cardiomyocytes, the sarco/endoplasmic reticulum (SER) is the site of several vital functions for the cell. It is the site for protein synthesis, post-translational modification and folding and it serves as a common route for transport of newly synthesized proteins to their destination (membrane proteins, secretory proteins). A proper balance between synthesis, maturation, and degradation of proteins is crucial for cell survival.
Other than cytoplasmic and mitochondrial proteins, translation of all other proteins occurs on ribosomes associated with the ER that provides a unique biochemical environment for proper protein folding. The ER provides the proper levels of Ca2+ concentration, a more oxidizing environment [40] and the required set of specialized enzymes and chaperones proteins to allow a level of extraordinary timely and efficient processing of newly synthesized proteins, quality control and secondary damage control.
An aminoacidic chain becomes functional only when it folds into its tertiary or quaternary structural form. Although the aminoacidic sequence of nascent proteins determines the subsequent levels of protein shape (secondary, tertiary, and quaternary structure), the spontaneous structural maturation of the proteins may be excessively slow and inefficient. Ubiquitous, highly conserved, specialized proteins, called chaperones, accompany and regulate the efficiency of protein folding.
Figure 4 summarizes the steps for protein modification following ribosomal synthesis. N-terminal hydrophobic signals are required to ensure the translation of proteins on the ER. Signal recognition particles ensure that the ribosomes are going to be locked to the ER and the N-terminal hydrophobic sequence will then fit into the lipid bilayer of the ER membrane. A signal peptidase will remove the hydrophobic sequence and the protein translation will continue in the ER. Several covalent modifications will be added to the nascent proteins including glycosylation, phosphorylation, γ-carboxylation, prenylation, and proteolysis among others. In the ER, the process of protein folding and post-translation modification is made temporarily and spatially possible by the catalyst function of the chaperone proteins. The peptidyl isomerase, the disulfide isomerase (PDI) and the Glucose-regulated proteins (GRP) in the ER assist the folding of nascent protein and the refolding of incorrectly folded or damaged proteins.

Illustrate the process of synthesis and maturation of secreted and membrane proteins. Ribosomes are targeted to the ER by signal recognition particles that recognize the N-terminal hydrophobic signals. The signal is removed in the ER and the translation and post-translational modification sequentially continue in the ER
Lacking clear understanding of their exact cellular function, chaperones are still best classified by their molecular weight. Within the major molecular chaperone families with biological interest in relation to myocardial diseases are the heat shock family proteins (HSPs) [41]. The Hsp70 proteins are the most widely studied and abundant in eukaryotic cells [42]. Recent studies indicated distinct functions among Hsp70 members [43]. They include a Ca2+-binding glucose-regulated class of stress proteins (GRP), which has a functional partnership with the Hsp90 chaperone family. The ER homolog of Hsp70, BiP (GRP78/BiP), and the co-chaperone GRP94 are induced by stimuli such as anoxia, glucose starvation and Ca2+ ionophores. They have been reported to show variable expression in the ischemic heart [44–46].
In addition to the chaperone molecules that increase the efficiency of the folding process, other folding catalyst proteins (disulfide isomerases) increase the rate of the folding steps. Stringent quality control systems in the ER ensure that only correctly folded proteins are sent to their final functional sites. The quality control is monitored by integrated signaling pathways that constantly monitor and adjust the levels of folding control. The quality control in the ER is integrated with other cellular compartments as well as with transcriptional and translational control to prevent cell toxicity by misfolded protein products [47].
In fact, molecular chaperones do not just regulate nascent protein folding, but can also rescue proteins from defects in folding or can redirect misfolded peptides toward the correct folded form (quality control—the unfolding protein response). Failure to fold correctly, to be refolded or to remain folded can give rise to cellular malfunctioning and to disease [47, 48]. We will first describe the role of ER as a signaling organelle and its role in the control of Ca2+ dynamics and signaling that, in turns, control vital functions and then the response of the ER when cells are subjected to acute and chronic environmental stimuli [49–51].
ER integrated signaling organelle
The ER is, in a simple vision, an intracellular tubular system that can appear as an inert compartment for transient Ca2+ storage. The ER however holds an essential role as Ca2+ regulator, and, more so the SR in cardiomyocytes, it coordinates several vital functions. In the cardiomyocytes, the SR is activated by electrical and chemical stimuli to provide a source of rapid exchange of Ca2+ while maintaining stable high levels of Ca2+ for protein biochemical processing. During the excitation–contraction coupling cycling the intracellular/intraSR concentration of Ca2+ oscillate of 10,000 times whereas, simultaneously, stably high level of [Ca2+]SR are required for the proper protein synthesis and post-translational processing.
In this the SR coordinates functions requiring different temporal resolution and quantity disparities of ions and elaborates signals integrated with other intracellular compartments. A spatial distribution of the signaling mediator Ca2+ and of Ca2+ related proteins (Ca2+-binding and Ca2+-regulated chaperones, Ca2+-pumps and transporters, Ca2+-channels) within the SR might be the trick to coordinate the different functions in the SR [52, 53]. The spatial distribution of Ca2+ handling proteins in regulating different cell functions has been studied in various cell types [54–57] and it can provide a controlled pool of Ca2+ in specialized SR subdomains. Thus, the heterogeneous spatial distribution of Ca2+ handling and binding proteins in SR functional subdomains will allow transient Ca2+ signaling for rhythmic alternation of contraction and relaxation and stable Ca2+ homeostasis for protein synthesis and maturation. Therefore, some parts of the SR enriched with Ca2+ pumps and receptors allow a more rapid oscillation of intraSR Ca2+ levels, whereas the differential distribution of Ca2+-binding proteins and chaperones will protect from vast oscillation allowing the proper biochemical environment for the different protein biochemistry functions. The spatial distribution of proteins within the SR microdomains and the integrated function of the SR Ca2+ homeostasis for the control of different cell function represent an important parameter in understanding the global malfunction in the myopathic heart, the general picture of the contractile dysfunction and the activation of death signaling pathways.
Ca2+ as integrated signaling molecule
Ca2+ is the mediator of the dual role of the ER and the key ion for cell function and life. Living organisms have evolved to accomplish complex biochemistry in a crowded aqueous environment where water solubility is a vital requirement. Organic molecules are required to be hydrophilic or hydrophilic coated, therefore charged, and among those, there are charged derivatives of phosphoric acid. Such derivatives compose many key molecules for life (ATP, nucleic acids). The low solubility of Ca2+-phosphate has selected mechanisms for Ca2+ extrusion and compartmentalization and evolved the use of this requirement to generate electrical and chemical gradients. Ca2+ homeostasis become crucial for many cell functions that include contraction–relaxation in muscle cells, protein synthesis, post-translation modification and folding, secretion, gene expression, cell cycle progression, and apoptosis.
[Ca2+]SR concentration is known to control the release of Ca2+ from the SR through the RyRs and therefore the SR Ca2+ load is a key self-level of control in cardiac contractile function. On the other hand, the free [Ca2+]SR controls the biochemistry processes of protein synthesis and modifications and chaperone interactions. SR resident Ca2+ binding and chaperone proteins Calreticulin and Calnexin seem to play a key role in mediating the sensing of [Ca2+]SR and the interaction with newly synthesized proteins as well as with other chaperones proteins to mediate protein folding in a Ca2+ dependent manner. High [Ca2+]SR has been shown to reduce the binding of Calreticulin to the chaperone protein PDI promoting the chaperones function of PDI. Under the same condition, the chaperone activity of Calreticulin is promoted. At low [Ca2+]SR as occurs during the release of Ca2+ from the SR the rate of protein synthesis, post-translational modification and secretion is vice-versa reduced. Thus, [Ca2+]SR is evolved to integrate various signaling from the transient control of contractile function of the myocytes to the control of protein synthesis, maturation, and quality control leading to a secondary signaling to the nucleus for a more sustained control of cell life.
Disruption of Ca2+ homeostasis and particularly SR Ca2+ balance produces the SR stress response. Upon persistence of SR stress and disruption of Ca2+ homeostasis, SR triggers a series of cells signals leading to cellular diseases and cell death. This is particularly important in the setting of pathological condition of disruption of SR Ca2+ homeostasis [58, 59].
ER stress, UPR, ERAD, and cell death
Various conditions can interfere with ER function and are collectively called ER stress, which in turn can be triggered by alterations of ER Ca2+ homeostasis [59]. The stress stimuli include (1) alteration in the redox environment in the ER, (2) inhibition of protein (N)—glycosylation leading to incorrect protein folding of nascent proteins that can foster protein denaturation and loss of function, (3) reduction in protein disulfide bonds, (4) expression of mutant proteins, and (5) [Ca2±]SR depletion.
Upon ER stress induction, a conserved stress response pathway, the Unfolding Protein Response (UPR) (Fig. 5) allows cells to tolerate accumulation of misfolded proteins and act to correct the defect [59]. In the UPR, a signal selectively activates the transcription of genes encoding for ER-localized proteins such as PDI to increase the rate of protein folding. GRPs are also upregulated to maintain or correct the protein folding state as well as protect folding intermediates from aggregating [60, 61]. Protein synthesis is inhibited by stimulation of kinases that inhibit the initiation events and terminally misfolded proteins are targeted to be destroyed by the addition of several ubiquitin residues that signals the polypeptide for degradation to the proteasome system, a process called ER-associated degradation (ERAD) [47, 62–64].

When unfolded proteins accumulate in the ER, a conserved eukaryotic stress response pathway (UPR) is activated. Ire1, residing in the ER membrane, appears to function as a sensor of the accumulation of unfolded proteins and activate the signal that enhances the transcription of chaperones genes. In eukaryotes, appropriate expression of chaperone genes requires regulatory elements termed ERSEs in their promoters. In addition to chaperone upregulation, several other signals emerge from the stressed ER including the induction of a growth/arrest cell death promoting transcription factor (CHOP). In addition protein synthesis is transiently downregulated because of phosphorylation of the eukaryotic initiation factor 2α by the kinase PKR-like ER kinase
The ER/SR itself contains resident proteins that act as the sensors to ER stress and activate the UPR signaling: the IRE1, a kinase and endoribonuclease composed of two subunits—α and β, the PERK kinase and the ATF6 transcription factor (α and β), a leucine zipper transcription factor. Through these sensors, alterations in the ER homeostasis affects different cellular functions and cell signaling to modify metabolism, gene expression, and apoptosis. The activation of the UPR occurs as a temporal sequence of events in response to the increase engagement of the chaperone protein BiP with the unfolded proteins in the ER (rev by [65]). Under basal conditions, BiP binds to the sensors preventing their activation. The binding of BiP to the unfolded proteins would shift the balance of BiP from the sensors and unengage them to be activated. Following ER stress, PERK undergo homodimerization and autophosphorylation. Phosphorylation and dimerization of PERK results in phosphorylation of the transcription factor elF2a and the blocking of protein translation by inhibiting the ribosomal assembly. The second activation in the UPR is the proteolitic cleavage of ATF6 and the release of its cytosolic domain. The ATF6 cleaved domain, translocate to the nucleus activating the transcription of genes for the ER chaperones proteins (BIP, PDI, and GRP94) to counteract the consequences of stressors. Given that the activation of this sensor will have to activate the transcription and translation of chaperones proteins, this response temporarily needs transcription and translation time. ATF4 transcription factor is also activated requiring the phosphorylation by elF2α thus being downstream of PERK. Finally, IRE1 initiate the transcription of XBP1 mRNA. The level of XBP1 is increased by ATF6 following the activation of the UPR. This third response is to activate the genes for the ERAD therefore this pathway is activated at last after the initiation of the UPR response during persistent ER stress.
Whereas the UPR and ERAD response are able to counteract a temporary stress condition, the UPR needs to be interrupted as much as is to be persevered while the stress induction persists. Negative and positive feedbacks are in place to account for those situations.
Two negative feed-back mechanisms repress PERKS by upregulation of GADD34, a member of the DNA-damage gene family, and its associated protein phosphatase 1 (PP1) and the p58IPK. The PP1 interrupt the PERK pathways by dephosphorylating elF2α whereas p58IPK directly dephosphorylates PERK.
The persistence of the UPR response, for counteracting chronic stress, is guaranteed by the self-induction of XBP1 gene by his protein product. The persistence of the retention of misfolded proteins activates the ERAD where the unfolded proteins recognized by chaperones proteins (BiP) are retrotranslocated in the cytosol via the Sec61 channel, polyubiquitinated, and directed to the proteasome for degradation. The UPR and the ERAD are partially overlapping mechanisms that normally eliminate misfolded proteins that are generated during the process of cell growth and differentiation, and even more in conditions of stress. Also the persistent parallel depletion of [Ca2+]ER/SR and the UPR initiate the pro-apoptotic pathways by several mechanisms.
Both PERK and ATF6 promotes apoptosis via activation of the transcription factor CHOP that suppresses the transcription of the pro-survival factor Bcl-2 [12]. Similar to the apoptotic extrinsic mechanism, the other UPR sensor IRE1 interact with Caspase 12 through the adaptor protein TRAF2 [66]. Upon stress, Caspase 12 dissociates from TRAF2 and activates apoptosis pathways. The activation of Caspase 12 though occurs by the cleavage by the Ca2+ dependant protease calpain [67]. Thus, once again increased [Ca2+]i, an ER stress factor and a well-known event in the failing myocardium, activates the calpain-induced Caspase 12 apoptotic pathways. Cellular Ca2+ overload activate apoptosis also by mitochondrial Ca2+ reuptake and inner-membrane potential damage associated with apoptosis.
A stimulus to trigger the UPR and the ERAD, is altered Ca2+ homeostasis and Ca2+ depletion from the ER lumen. Ca2+ gradients in the ER are generated by SR Ca2+-ATPase (SERCA) in many cell types [68]. Studies focusing on the regulation of ER Ca2+-ATPase under conditions evoking the UPR found a correlation between Ca2+ depletion and SERCA2b mRNA in neurons [69]. As mentioned, alteration in Ca2+ homeostasis in failing hearts has been shown to be a key determinant of contractile dysfunction [14, 70] from the myocyte level to the entire organ. Changes in diastolic intracellular Ca2+ have been shown to be deleterious for cell survival activating kinases, caspases, proteases, and apoptotic pathways that induces myofilaments degradation. The parallel chronic depletion in the [Ca2+]ER add further mechanisms for the deleterious outcome for cell function and cell survival. A systematic study of cell death in the failing human hearts [71] underlined the contemporary presence of several mechanisms of cell death in the failing myocardium: apoptosis, oncosis and autophagic cell death. This report describes the myocyte cellular degeneration in human heart failure underlying the presence of a defect of balance between ubiquitination and degradation in heart failure following stress and oxidative damage.
Conclusions
The ER Ca2+ homeostasis and the tight control of SR Ca2+ levels emerge as key factors for the cardiomyocytes contractile function as well as the cell vital function. The ER integrates the signaling for the control of the rhythmic alternation of contraction and relaxation as well as the cell survival signaling. Disruption of the homeostasis of Ca2+ and of the SR Ca2+ homeostasis results in disastrous consequences for the contractile function, in ER stress and in overall devastating consequences for cell function and survival (Fig. 6).

Disruption of SR and cell Ca2+ homeostasis and changes in the cellular architecture are well-recognized features of the failing myocardium. Consequences of abnormal Ca2+ in the subcellular compartments has deleterious consequences on the contractile function of the cardiomyocytes and on the ER protein biochemistry and cell death signaling
References
Gwathmey JK, Copelas L, MacKinnon R et al (1987) Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res 61:70–76
Gwathmey JK, Hajjar RJ (1990) Relation between steady-state force and intracellular [Ca2+] in intact human myocardium. Index of myofibrillar responsiveness to Ca2+. Circulation 82:1266–1278
Bers DM (2002) Cardiac excitation–contraction coupling. Nature 415:198–205
Gwathmey JK, Slawsky MT, Hajjar RJ, Briggs GM, Morgan JP (1990) Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J Clin Invest 85:1599–1613
Hasenfuss G, Pieske B (2002) Calcium cycling in congestive heart failure. J Mol Cell Cardiol 34:951–969
Bers DM (2000) Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res 87:275–281
Bers DM (2002) Sarcoplasmic reticulum Ca release in intact ventricular myocytes. Front Biosci 7:d1697–d1711
Gwathmey JK, Hajjar RJ (1990) Intracellular calcium related to force development in twitch contraction of mammalian myocardium. Cell Calcium 11:531–538
Piacentino V III, Weber CR, Chen X et al (2003) Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res 92:651–658
Ke Y, Wang L, Pyle WG, de Tombe PP, Solaro RJ (2004) Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ Res 94:194–200
Communal C, Sumandea M, de Tombe P, Narula J, Solaro RJ, Hajjar RJ (2002) Functional consequences of caspase activation in cardiac myocytes. Proc Natl Acad Sci USA 99:6252–6256
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21:1249–1259
Brittsan AG, Ginsburg KS, Chu G et al (2003) Chronic SR Ca2+-ATPase inhibition causes adaptive changes in cellular Ca2+ transport. Circ Res 92:769–776
Beuckelmann DJ, Erdmann E (1992) Ca(2+)-currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Res Cardiol 87(Suppl 1):235–243
Meyer M, Schillinger W, Pieske B et al (1995) Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92:778–784
Hasenfuss G, Reinecke H, Studer R et al (1996) Calcium cycling proteins and force–frequency relationship in heart failure. Basic Res Cardiol 91(Suppl 2):17–22
Schillinger W, Janssen PM, Emami S et al (2000) Impaired contractile performance of cultured rabbit ventricular myocytes after adenoviral gene transfer of Na(+)–Ca(2+) exchanger. Circ Res 87:581–587
Schmidt U, Hajjar RJ, Helm PA, Kim CS, Doye AA, Gwathmey JK (1998) Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol 30:1929–1937
Schmidt U, Hajjar RJ, Kim CS, Lebeche D, Doye AA, Gwathmey JK (1999) Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. Am J Physiol 277:H474–H480
Schillinger W, Lehnart SE, Prestle J et al (1998) Influence of SR Ca(2+)-ATPase and Na(+)-Ca(2+)-exchanger on the force–frequency relation. Basic Res Cardiol 93(Suppl 1):38–45
Schmidt U, Hajjar RJ, Helm PA, Kim CS, Doye AA, Gwathmey JK (1998) Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol 30:1929–1937
Brixius K, Savvidou-Zaroti P, Mehlhorn U, Bloch W, Kranias EG, Schwinger RH (2002) Increased Ca2+-sensitivity of myofibrillar tension in heart failure and its functional implication. Basic Res Cardiol 97(Suppl 1):I111–I117
Pieske B, Maier LS, Bers DM, Hasenfuss G (1999) Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ Res 85:38–46
Pieske B, Schlotthauer K, Schattmann J et al (1997) Ca(2+)-dependent and Ca(2+)-independent regulation of contractility in isolated human myocardium. Basic Res Cardiol 92(Suppl 1):75–86
Schmidt U, Hajjar RJ, Kim CS, Lebeche D, Doye AA, Gwathmey JK (1999) Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. Am J Physiol 277:H474–H480
Winslow RL, Rice J, Jafri S (1998) Modeling the cellular basis of altered excitation–contraction coupling in heart failure. Progr Biophys Mol Biol 69:497–514
Lehnart SE, Schillinger W, Pieske B, Prestle J, Just H, Hasenfuss G (1998) Sarcoplasmic reticulum proteins in heart failure. Ann NY Acad Sci 853:220–230
Hajjar RJ, Schmidt U, Matsui T et al (1998) Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci USA 95:5251–5256
Frank KF, Bolck B, Brixius K, Kranias EG, Schwinger RH (2002) Modulation of SERCA: implications for the failing human heart. Basic Res Cardiol 97(Suppl 1):I72–I78
Haghighi K, Kolokathis F, Pater L et al (2003) Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 111:869–876
Schmitt JP, Kamisago M, Asahi M et al (2003) Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299:1410–1410
Ahmad Z, Green FJ, Subuhi HS, Watanabe AM (1989) Autonomic regulation of type 1 protein phosphatase in cardiac muscle. J Biol Chem 264:3859–3863
Gupta RC, Neumann J, Watanabe AM, Lesch M, Sabbah HN (1996) Evidence for presence and hormonal regulation of protein phosphatase inhibitor-1 in ventricular cardiomyocyte. Am J Physiol 270:H1159–H1164
Neumann J, Eschenhagen T, Jones LR et al (1997) Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol 29:265–272
Carr AN, Schmidt AG, Suzuki Y et al (2002) Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol 22:4124–4135
Yue TL, Gu JL, Wang C et al (2000) Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J Biol Chem 275:37895–37901
Hasenfuss G, Schillinger W, Lehnart SE et al (1999) Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation 99:641–648
Pogwizd SM, Bers DM (2004) Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med 14:61–66
Shannon TR, Pogwizd SM, Bers DM (2003) Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res 93:592–594
van den Berg B, Ellis RJ, Dobson CM (1999) Effects of macromolecular crowding on protein folding and aggregation. Embo J 18:6927–6933
Benjamin IJ, McMillan DR (1998) Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ Res 83:117–132
Tavaria M, Gabriele T, Kola I, Anderson RL (1996) A hitchhiker’s guide to the human Hsp70 family. Cell Stress Chaperones 1:23–28
James P, Pfund C, Craig EA (1997) Functional specificity among Hsp70 molecular chaperones. Science 275:387–389
Gething MJ, Sambrook J (1992) Protein folding in the cell. Nature 355:33–45
Nishizawa J, Nakai A, Higashi T et al (1996) Reperfusion causes significant activation of heat shock transcription factor 1 in ischemic rat heart. Circulation 94:2185–2192
Glass MG, Fuleihan F, Liao R et al (1993) Differences in cardioprotective efficacy of adrenergic receptor antagonists and Ca2+ channel antagonists in an animal model of dilated cardiomyopathy. Effects on gross morphology, global cardiac function, and twitch force. Circ Res 73:1077–1089
Gorza L, F dM (2005) Protein unfolding in cardiomyopathies. Heart Failure Clinic 1:237–250
Chevet E, Cameron PH, Pelletier MF, Thomas DY, Bergeron JJ (2001) The endoplasmic reticulum: integration of protein folding, quality control, signaling and degradation. Curr Opin Struct Biol 11:120–124
Ellis RJ, Hemmingsen SM (1989) Molecular chaperones: proteins essential for the biogenesis of some macromolecular structures. Trends Biochem Sci 14:339–342
Ellis RJ, Hartl FU (1996) Protein folding in the cell: competing models of chaperonin function. Faseb J 10:20–26
Hartl FU (1996) Molecular chaperones in cellular protein folding. Nature 381:571–579
Pezzati R, Bossi M, Podini P, Meldolesi J, Grohovaz F (1997) High-resolution calcium mapping of the endoplasmic reticulum-Golgi-exocytic membrane system. Electron energy loss imaging analysis of quick frozen-freeze dried PC12 cells. Mol Biol Cell 8:1501–1512
Papp S, Dziak E, Michalak M, Opas M (2003) Is all of the endoplasmic reticulum created equal? The effects of the heterogeneous distribution of endoplasmic reticulum Ca2+-handling proteins. J Cell Biol 160:475–479
Belan PV, Gerasimenko OV, Berry D, Saftenku E, Petersen OH, Tepikin AV (1996) A new technique for assessing the microscopic distribution of cellular calcium exit sites. Pflugers Arch 433:200–208
Petersen OH, Burdakov D, Tepikin AV (1999) Polarity in intracellular calcium signaling. Bioessays 21:851–860
Petersen OH, Burdakov D, Tepikin AV (1999) Regulation of store-operated calcium entry: lessons from a polarized cell. Eur J Cell Biol 78:221–223
Johnson JD, Chang JP (2000) Function- and agonist-specific Ca2+ signalling: the requirement for and mechanism of spatial and temporal complexity in Ca2+ signals. Biochem Cell Biol 78:217–240
LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 3:862–872
Verkhratsky A, Toescu EC (2003) Endoplasmic reticulum Ca(2+) homeostasis and neuronal death. J Cell Mol Med 7:351–361
Liu H, Bowes RC III, van de Water B, Sillence C, Nagelkerke JF, Stevens JL (1997) Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J Biol Chem 272:21751–21759
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233
Ciechanover A, Elias S, Heller H, Ferber S, Hershko A (1980) Characterization of the heat-stable polypeptide of the ATP-dependent proteolytic system from reticulocytes. J Biol Chem 255:7525–7528
Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA (2004) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429:841–847
Spiro RG (2004) Role of N-linked polymannose oligosaccharides in targeting glycoproteins for endoplasmic reticulum-associated degradation. Cell Mol Life Sci 61:1025–1041
Rutkowski DT, Kaufman RJ (2004) A trip to the ER: coping with stress. Trends Cell Biol 14:20–28
Yoneda T, Imaizumi K, Oono K et al (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 276:13935–13940
Nakagawa T, Zhu H, Morishima N et al (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403:98–103
Ashby MC, Tepikin AV (2001) ER calcium and the functions of intracellular organelles. Semin Cell Dev Biol 12:11–17
Verkhratsky A, Petersen OH (2002) The endoplasmic reticulum as an integrating signalling organelle: from neuronal signalling to neuronal death. Eur J Pharmacol 447:141–154
del Monte F, Hajjar RJ (2003) Targeting calcium cycling proteins in heart failure through gene transfer. J Physiol 546:49–61
Kostin S, Pool L, Elsasser A et al (2003) Myocytes die by multiple mechanisms in failing human hearts. Circ Res 92:715–724
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
del Monte, F., Hajjar, R.J. Intracellular devastation in heart failure. Heart Fail Rev 13, 151–162 (2008). https://doi.org/10.1007/s10741-007-9071-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-007-9071-9