
Abstract
A number of epidemiological and animal studies have suggested a cardioprotective role for estrogen. This review will focus on the cardioprotective role of estrogen in ischemia-reperfusion injury. Estrogen binding to receptors can lead to altered gene expression and estrogen has been shown to induce expression of a number of genes that have been suggested to be important in cardioprotection. Estrogen is reported to increase expression of the plasma membrane glucose transporter GLUT4 and to increase carbohydrate metabolism. Estrogen has also been reported to increase mitochondrial biogenesis and to alter mitochondrial generation of reactive oxygen species. Estrogen results in upregulation of cardiac eNOS and nNOS, which have been shown previously to be important mediators of cardioprotection. Nitric oxide has been shown to result in S-nitrosylation and inhibition of the L-type calcium channel, thereby reducing calcium loading during ischemia. Nitric oxide has also been reported to inhibit complex I and inhibition of complex I has been reported to reduce activation of the mitochondrial permeability transition pore. Nitric oxide has been shown to result in activation of the mitochondrial KATP channel, which has been shown to be involved in cardioprotection. Estrogen can also activate rapid non-genomic pathways that activate cardioprotective-signaling pathways such as the phosphatidylinositol-3-kinase (PI-3 kinase) pathway which has also been shown to initiate protection. Taken together, estrogen by genomic and non-genomic pathways can result in the initiation of a number of signaling pathways that enhance cardioprotection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epidemiological studies have suggested that pre-menopausal females have a reduced incidence of cardiovascular disease [1–3]. Much of the cardiovascular protection observed in females have been attributed to beneficial effects of estrogen on lipid profile and endothelial function [4]. The direct effects of estrogen on cardiac myocytes have not been extensively studied. Furthermore, despite reduced cardiovascular disease in pre-menopausal females, in a large clinical trial, hormone replacement therapy did not reduce cardiovascular disease [5]. The lack of estrogen mediated protection in the Women’s Health Initiative (WHI) contrast not only with prior epidemiological studies, but also with data from animal studies. Potential reasons for the lack of protection in the WHI study, such as potential differences in the pharmacology between conjugated equine estrogen and 17-β-estradiol and the age of women at the start of treatment have been discussed in detail by others [6–8]. This review will focus on the data in animal models showing a protective effect of estrogen. Perhaps a better understanding of the mechanisms by which estrogen mediates protection in animal studies will provide insight into why hormone replacement therapy was not protective in the WHI study.
Estrogen receptor signaling
Most of the action of estrogen has been attributed to estrogen binding to either estrogen receptor (ER)-α or ER-β, two nuclear hormone receptors that act as ligand-activated transcription factors binding to DNA response elements. ER-α and ER-β are differentially expressed in different tissues of the body. ER-α and ER-β activate some common genes, but they also each activate a unique set of genes. Furthermore, ER-α and ER-β can oppose each other in a ying-yang relationship [9]. For example, ER-α induces and ER-β represses expression of apolipoprotein E in the hippocampus [10]. ER-α and ER-β, which can form homo and heterodimers, bind to DNA resulting in the recruitment of co-activators and co-repressors that modify estrogen mediated transcription. The expression of co-activators and co-repressors is tissue-specific and depends on cell context which will modify the effects of estrogen in a tissue specific manner. For example mitogen activated protein kinases (MAPK) can phosphorylate these co-regulators and modify their activity. Details regarding the complexity of ER interaction with co-regulators have been reviewed elsewhere [6, 11]. In addition, ligand bound ER can bind to DNA indirectly through complexes in association with SP1 (stimulator protein 1) or AP1 (activator protein 1) [12]. IGF and EGF have also been reported to cross talk with ER gene expression. IGF-1 can increase uterine weight in ovariectomized mice by a pathway that requires ER-α, since it does not occur in mice lacking ER-α [13]. Cross talk between ER and other transcription factors such as nuclear factor kappa B has also been reported [14].
In addition to estrogen action via binding to ER and activation of gene expression, there are data suggesting that estrogen can bind to an ER localized to the plasma membrane and acutely activate PI3-kinase and other signaling pathways [15]. There are also data suggesting that estrogen can bind to and signal via a G protein coupled receptor (GPR 30) [16]. Thus depending on the relative mix of receptors that are present in a target tissue, the response of the tissue to estrogen can differ. Additional diversity is provided by polymorphisms in ER as well as post-translational modification of ER such as S-nitrosylation and N-acetyl-glycosylation.
Estrogen and cardioprotection in animal models
A number of different models and approaches have been used to examine the effect of estrogen on ischemia-reperfusion injury. One approach has been to examine whether there are male–female differences in ischemia-reperfusion injury. Another approach is to examine the effect of addition of exogenous estrogen.
Male–female differences in I/R
Some studies have reported that females have reduced ischemia-reperfusion injury [17, 18]. Other studies have failed to observe a male–female difference in ischemia-reperfusion injury [19–21]. However, females have reduced ischemia-reperfusion injury in a number of transgenic mouse models characterized by increased contractility and in wild-type hearts with addition of isoproterenol or elevated extracellular calcium [21–26]. These data raise the possibility that the discrepancies regarding endogenous protection in females may be related to the sympathetic tone or contractile state in the different models.
Treatment with estrogen
There also are many studies showing that acute treatment of animals or perfused hearts with exogenous estrogen can reduce ischemia-reperfusion injury [20, 27–30]. Hale et al. reported that bolus IV administration of beta estradiol (10 μg), but not administration of 1 mg of alpha estradiol (which does not bind to ER) reduced infarct size in rabbits [29]. Booth et al. [30] showed that treatment of rabbits with 20 μg of estradiol prior to coronary occlusion reduced infarct size (19%) compared to vehicle (48%). Similarly, Das and Sarkar reported [28] that pretreatment of rabbits with estradiol (10 μg/kg iv) prior to coronary artery ligation significantly reduced infarct size (19% versus 40%). Sbarouni et al. showed that 4 week treatment with estrogen reduces infarct size in oophorectomized female rabbits on a normal [31], and cholesterol-enriched diet [32]. However, raloxifene treatment did not reduce infarct size [32]. It has also been reported that the inhibitors of the mitochondrial KATP channel block the protection afforded by estrogen [33]. Taken together the data suggest that estrogen can be protective.
Which ER is involved and are the effects via gene expression or acute signaling?
As discussed above, estrogen mediates most of its effect by binding to the estrogen receptor. A number of studies have examined whether the protection afforded by estrogen is mediated by ER-α or ER-β. These studies have been carried out using either genetically altered mice that lack either ER-α or ER-β or by addition of an ER-α or ER-β selective agonist. Unfortunately there is no consensus regarding which estrogen receptor mediates protection against ischemia-reperfusion injury. There are data suggesting a role for both ER-α [18, 27, 34] and ER-β [25, 35–37] in mediating cardioprotection. Possible reasons for the discrepancy include different models of ischemia-reperfusion and different end-points. Another variable, particularly in the studies using ER-α and ER-β agonists, is the dose and the timing of the addition of the agonist. In some studies, high levels of an ER-α agonist given for a short time were protective [27]; this protection may be mediated by acute-signaling pathways rather than gene induction. Another study showed protection by addition of an ER-β agonist after long-term (2 week) addition [36]; the protection observed in this study is likely mediated by altered gene expression. Thus both ER-α and ER-β may mediate protection, but by different mechanisms. Furthermore, altered gene expression is likely to be important, but acute effects of estrogen may also be involved and these acute and chronic effects may be mediated by different estrogen receptors. Also, some of the effects of estrogen are associated with high non-physiological levels and the mechanisms involved may not be mediated by binding to ER.
Estrogen mediated mechanisms of cardioprotection
Regardless of which ER mediates the protection, the mechanisms by which estrogen elicits cardioprotection in females are poorly understood. Given the lack of protection observed by HRT in the WHI, it is important to better understand the mechanisms by which estrogen might elicit cardioprotection. In this review we will focus on the role of estrogen and its well-established target nitric oxide synthase in cardioprotection. There are considerable data suggesting that alterations in nitric oxide synthase expression and signaling are important for protection in females [24, 38–40]. Altered expression of other genes are also likely to be involved and we will also review estrogen-mediated changes in gene expression that might play a role in cardioprotection.
Nitric oxide and cardioprotection in females
It has been well-established that estrogen results in increased expression of several nitric oxide synthase (NOS) isoforms [4, 39, 40]. The increase in basal NOS levels can lead to an increase in baseline nitric oxide generation in females [38, 40]. The increase in NOS in females has been shown to result in improved endothelial function. An increase in eNOS, mediated by ER-β, has been reported in cardiac myocytes [39]. Chen et al. have reported an increase in nNOS in cardiac myocytes [41] and Sun et al. have reported an increase in eNOS in the heart associated with caveolin 3 [40], the cardiomyocyte specific caveolin. The increase in eNOS and nNOS in cardiomyocytes in females is interesting in light of the well established role for increased NOS in cardioprotection [42]. Nitric oxide is suggested to mediate expression of cyclooxygenase 2 (COX-2), which has also been shown to be involved in cardioprotection [42]. Furthermore an increase in nitric oxide has been shown to be involved in cardioprotection via activation of protein kinase G (PKG), which leads to activation of mitochondrial pathways including activation of an ATP regulated mitochondrial channel that allows transport of K+ into the mitochondria (mito KATP channel) [43]. Opening of the mitochondrial KATP channel has been reported to induce cardioprotection. An increase in nitric oxide has also been shown to result in increased S-nitrosylation of complex I of mitochondria [44]; it is suggested that this might alter ROS generation during ischemia and/or reperfusion. An increase in NOS and nitric oxide in females has also been shown to cause increased S-nitrosylation of the L-type Ca2+ channel, which results in less Ca2+ loading during ischemia [40]. There are also a number of studies showing that nitric oxide can alter cell metabolism [45–47]. Nitric oxide is also reported to increase mitochondrial biogenesis and reduce mitochondrial generation of ROS [47]. An increase in NOS in females can thus alter gene expression and activate acute nitric oxide signaling pathways. Given the pleiotropic effects of nitric oxide, it is likely that the increased NOS in female cardiomyocytes has an important role in the male-female differences in cardioprotection.
Estrogen regulated gene expression
There are a large number of genes which are regulated by estrogen in a tissue specific manner. Otsuki et al. examined gene changes in hearts from ovariectomized females treated for 3 weeks with estradiol compared to vehicle [48]. They reported an induction of seven genes and decreased expression of nine genes [48]. The induced genes included lipocalin-type prostaglandin D synthase and dipeptidase I. The repressed genes included thymosin beta10 and several types of procollagen. Gabel el al. performed gene profiling to determine genes that are differentially expressed in hearts from mice lacking ER-β (compared to WT and αERKO mice) [25]. Loss of ER-β was found to lead to an induction of solute carrier 4 (member 1) and decreased expression of a number of metabolism genes including SPOT14 homolog, lipoprotein lipase, ATP citrate lyase, stearoyl CoA desaturase and fatty acid synthase [25]. These data suggest that estrogen via ER-β results in induction of a number of genes involved in metabolism. The effect of an increase in these genes by ER-β is unclear but worthy of study. It is interesting that mice lacking eNOS were also reported to have an increase in solute carrier family 4 (member 1) and a decrease in stearoyl-CoA desaturase [49]. A cardiac specific stearoyl CoA desaturase has been recently reported [50]. Stearoyl CoA desaturase catalyzes the synthesis of monounsaturated fatty acids from saturated fatty acids; monounsaturated fatty acids are important components in membrane phospholipids. As reviewed elsewhere [51] leptin has been reported to repress hepatic stearoyl CoA desaturase and expression of stearoyl CoA desaturase is increased in leptin deficiency. Mice with global deletion of stearoyl CoA desaturase have decreased body fat. It is therefore not clear that increased levels of this enzyme per se would enhance cardioprotection in females, although the role of this enzyme in cardiac and hepatic tissue could be different. ATP citrate lyase converts citrate to oxaloacetate and acetyl CoA; acetyl CoA is then converted to malonyl CoA which is an inhibitor of carnitine-palmitoyl transferase 1 (CPT-1) and thereby inhibits fatty acid oxidation. Interestingly, many of the genes decreased in the βERKO females are regulated by sterol receptor element binding protein (SREBP). SREBP-1α is reported to bind directly to the ERE(1/2) motifs and enhance ER binding when both ER subtypes are present [52]. A recent study by Nikolic et al. [36] examined cardiac genes induced by treatment of ovariectomized females with an ER-β selective agonist, DPN (2,3-bis(4-hydroxyphenyl)-propionitrile). DPN was reported to increase expression of over 100 genes, including COX2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase, an enzyme important for regulating glycolysis [53].
Other studies have used a candidate gene approach and have identified a number of genes regulated (directly or indirectly) by estrogen, including, peroxisome proliferator-activated receptor gamma-coactivator 1α (PGC-1α), connexin 43 [54, 55], adenine nucleotide translocator [56], heat shock proteins [57], mitochondrial complex IV [58], GLUT4 [59], and MCIP1 an inhibitor of calcineurin [60]. Many of these proteins have been suggested to be important in cardioprotection [61, 62].
Estrogen and metabolism
As discussed, ER-β increases the level of several key enzymes involved in substrate selection. In addition to altered regulation of metabolism genes, estrogen could regulate metabolism by alterations in signaling pathways such as nitric oxide signaling which is also upregulated in females, due to induction of NOS. Since estrogen results in altered expression of a large number of metabolism genes, coupled with the estrogen induction of NOS which can also regulate metabolism, the relationship between estrogen and metabolism and its potential role in cardioprotection will be discussed.
Mitochondria and mitochondrial biogenesis
Mitochondria from females are reported to have increased maximum rates of electron transport and increased oxygen consumption [63]. Females also have increased mitochondrial biogenesis and reduced generation of mitochondrial ROS [64]. However, there are also reports that estrogen can increase mitochondrial ROS generation resulting in activation of redox sensitive transcription factors. Mitochondrial generation of ROS is complex and may depend on the levels of estrogen as well as other signaling pathways that are activated in the cell. Additional studies will be needed to resolve the role of estrogen on mitochondria and ROS generation. Increased mitochondrial generation of ROS has been a popular theory of aging. It has been suggested that with aging there is increased ROS mediated damage to electron transport chain components, which causes increased ROS production leading to a downward spiral. It has also been suggested that the increased longevity in females is related to altered mitochondrial electron transport and reduced ROS [64, 65]. NO is reported to be involved in increased mitochondrial biogenesis and increased longevity associated with caloric restriction [47, 66]. Nisoli et al. also reported that an increase in NO results in an increase in Sirt1 [66], a transcription factor associated with an increase in longevity and cardioprotection [67]. It is tempting to speculate that perhaps these mitochondrial alterations in females are mediated by nitric oxide.
Consistent with reduced ROS damage in females, Yan et al. using a proteomics approach reported age-dependent differences between male and female monkey hearts in glycolytic and mitochondrial electron-transport pathways [68]. They note that the changes in the old male monkeys are similar to changes that occur in disease and suggest that the lack of these changes in old female hearts is consistent with delayed cardiovascular risk in females. Also consistent with estrogen mediated changes in mitochondrial proteins, Stirone et al. [65] have shown that in blood vessels, estrogen increases levels of the nuclear coded cytochrome c, subunit IV of complex IV and manganese SOD, and increases the level of subunit I of complex IV that is coded in the mitochondria. Stirone et al. [65] further showed that hydrogen peroxide levels were decreased in estrogen treated animals. These changes were inhibited by the ER antagonist ICI-182, 780, but not by inhibitors of nitric oxide or the PI3-kinase pathway. Mitochondrial DNA contains estrogen receptor response elements. ER has been detected in the mitochondria [69, 70]; however these data are controversial [71].
Diabetes and body weight
A number of studies have suggested a link between estrogen and metabolism. Data from the Heart and Estrogen/Progestin Replacement Study (HERS) showed that there was significantly less type II diabetes in women on HRT [72]. Furthermore, the αERKO mice develop type II diabetes and exhibit an increase in body fat [73]. Interestingly ovariectomy of αERKO mice decreases body fat and body weight [74]. A human male lacking ER-α was also reported to have glucose intolerance [75], suggesting that the phenotype observed in mice is also relevant to humans. Whether the increase in type II diabetes associated with loss of ER-α is due to lack of ER-α signaling or due to unopposed ER-β signaling is unresolved. It is also interesting that eNOS-KO mice were reported to have an increase in body weight [47], suggesting a possible link between estrogen, NOS and altered body weight and metabolism. Thus there are considerable data suggesting that estrogen can regulate whole body metabolism; however, there are very limited data examining the effect of estrogen on myocardial metabolism.
Cardiac metabolism
We have recently found that substrate selection in heart is different in males versus females [25]. Using 13C NMR, labeled fatty acid, and carbohydrates, and measuring incorporation of the label into the C4 of glutamate, Gabel et al. [25] reported that in the C4 of glutamate, the ratio of 13C label from carbohydrate relative to fatty acid was significantly higher in females compared to males. The increase in carbohydrate relative to fatty acid utilization in females is consistent with the estrogen induction of ATP citrate lyase and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. How might this alteration in substrate selection result in cardioprotection? Numerous studies have shown that stimulation of glucose oxidation is protective. It has been proposed that increasing glucose oxidation during ischemia-reperfusion will result in pyruvate oxidation by pyruvate dehydrogenase (PDH) rather than pyruvate conversion to lactate; the latter can increase acidosis and result in calcium overload during ischemia and early reperfusion [76, 77]. Drugs that increase glucose oxidation, such as activators of PDH, have been shown to reduce ischemia-reperfusion injury [78, 79]. An increase in glucose oxidation in females might therefore be cardioprotective. In contrast to the observation by Gabel et al. [25] that females have increased glucose oxidation relative to fatty acid oxidation, Saeedi et al. [80] reported that relative to males, females have reduced glucose oxidation; however, in this study females also exhibited significantly poorer recovery of function than males. The reason for this discrepancy, particularly the enhanced ischemia-reperfusion injury in females is unclear.
In contrast to the concordant increase in mitochondrial biogenesis by nitric oxide and estrogen, an increase in nitric oxide is suggested to decrease carbohydrate oxidation and increase fatty acid oxidation, an effect opposite of that suggested for estrogen by Gabel el al. It has been shown in an in vivo model that acute inhibition of NOS causes an increase in glucose oxidation in heart [45, 46] This would suggest that NO results in a decrease in glucose oxidation. Since females have an increase in NO, if the metabolic effects observed in females are due solely to the effect of NO, one would expect a decrease in glucose oxidation in females. This suggests that perhaps estrogen alters expression of genes involved in metabolism resulting in increased glucose oxidation, but at the same time estrogen increases NO which counters the gene changes. It is also interesting that in a perfused heart model, inhibition of NOS does not cause an increase in carbohydrate metabolism [81]. Nitric oxide is also reported to alter glucose metabolism in the heart, but the results are somewhat conflicting. Lei et al. report that exogenous nitric oxide reduces glucose transporter translocation in heart [82]. However, Li et al. report that AMP kinase stimulation of GLUT4 translocation is at least partially mediated by an increase in NO [83]. Additional experiments will be necessary to unravel the complex role of estrogen and NO in metabolism.
Summary and conclusions
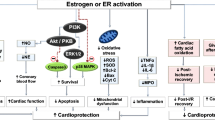
Estrogen binding to receptors can lead to altered gene expression, which can alter the response of the cell to ischemia-reperfusion. Figure 1 illustrates mechanisms by which estrogen might mediate cardioprotection. Nitric oxide synthase isoforms have been shown in many tissues to be increased in response to estrogen. Estrogen also results in altered expression of many additional genes. As discussed elsewhere, estrogen mediated gene expression is different in different tissues and can vary depending on co-repressors, co-activators, and other signaling pathways active in the tissue. We are just beginning to identify how estrogen alters cardiac gene expression. How does this all result in protection in females? Estrogen results in upregulation of cardiac eNOS and nNOS which have been shown previously to be important mediators of cardioprotection. An increase in NO has been shown in females to increase S-nitrosylation of the L-type Ca channel and thereby reduce Ca loading during ischemia and reperfusion [40]. An increase in NO has also been reported to enhance cardioprotection by activation of the mitochondrial KATP channel [84]. Estrogen also induces expression of a number of other genes that have been suggested to be important in cardioprotection.

The figure illustrates mechanisms involved in estrogen-mediated cardioprotection. Estrogen alters expression of a number of cardioprotective genes such as nitric oxide synthase (NOS). An increase in NOS activity results in stimulation of nitric oxide (NO) production, which can activate the mitochondrial KATP channel [84]. NO can also lead to S-nitrosylation of the L-type calcium channel [40] which would reduce calcium loading and thereby reduce opening of the mitochondrial permeability transition pore (mPTP). NO also results in S-nitrosylation of complex I of the mitochondria [44], which inhibits opening of the mitochondrial permeability transition pore (mPTP) [85]. NO also inhibits cytochrome c oxidase which would reduce Δψ and thus reduce calcium uptake into the mitochondria and opening of the mPTP. Estrogen is also reported to reduce mitochondrial generation of reactive oxygen species (ROS) [65] which would also reduce mPTP. Furthermore estrogen increases glucose oxidation, which, during ischemia-reperfusion, would reduce lactate production and thus reduce cytosolic increases in sodium and calcium. Finally estrogen can act via rapid non-genomic mechanisms to increase signaling pathways, such as the PI3-kinase pathway, to enhance protection in females
References
Barrett-Connor E (1997) Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys Lecture. Circulation 95:252–264
Crabbe DL, Dipla K, Ambati S, Zafeiridis A, Gaughan JP, Houser SR, Margulies KB (2003) Gender differences in post-infarction hypertrophy in end-stage failing hearts. J Am Coll Cardiol 41:300–306
Hayward CS, Kelly RP, Collins P (2000) The roles of gender, the menopause and hormone replacement on cardiovascular function. Cardiovasc Res 46:28–49
Mendelsohn ME, Karas RH (1999) The protective effects of estrogen on the cardiovascular system. N Engl J Med 340:1801–1811
Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J (2002) Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. Jama 288:321–333
Mendelsohn ME, Karas RH (2005) Molecular and cellular basis of cardiovascular gender differences. Science 308:1583–1587
Turgeon JL, Carr MC, Maki PM, Mendelsohn ME, Wise PM (2006) Complex actions of sex steroids in adipose tissue, the cardiovascular system, and brain: Insights from basic science and clinical studies. Endocr Rev 27:575–605
Ouyang P, Michos ED, Karas RH (2006) Hormone replacement therapy and the cardiovascular system lessons learned and unanswered questions. J Am Coll Cardiol 47:1741–1753
Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C (2003) Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol Endocrinol 17:203–208
Wang JM, Irwin RW, Brinton RD (2006) Activation of estrogen receptor alpha increases and estrogen receptor beta decreases apolipoprotein E expression in hippocampus in vitro and in vivo. Proc Natl Acad Sci USA 103:16983–16988
Deroo BJ, Korach KS (2006) Estrogen receptors and human disease. J Clin Invest 116:561–570
Sanchez R, Nguyen D, Rocha W, White JH, Mader S (2002) Diversity in the mechanisms of gene regulation by estrogen receptors. Bioessays 24:244–254
Curtis SW, Washburn T, Sewall C, DiAugustine R, Lindzey J, Couse JF, Korach KS (1996) Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci USA 93:12626–12630
Kalaitzidis D, Gilmore TD (2005) Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol Metab 16:46–52
Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK (2000) Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407:538–541
Filardo EJ, Thomas P (2005) GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab 16:362–367
Bae S, Zhang L (2005) Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts: focus on Akt and protein kinase C signaling. J Pharmacol Exp Ther 315:1125–1135
Wang M, Crisostomo P, Wairiuko GM, Meldrum DR (2006) Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol 290:H2204–2209
Przyklenk K, Ovize M, Bauer B, Kloner RA (1995) Gender does not influence acute myocardial infarction in adult dogs. Am Heart J 129:1108–1113
Li Y, Kloner RA (1995) Is There a Gender Difference in Infarct Size and Arrhythmias Following Experimental Coronary Occlusion and Reperfusion? J Thromb Thrombolysis 2:221–225
Cross HR, Lu L, Steenbergen C, Philipson KD, Murphy E (1998) Overexpression of the cardiac Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circ Res 83:1215–1223
Cross HR, Kranias EG, Murphy E, Steenbergen C (2003) Ablation of PLB exacerbates ischemic injury to a lesser extent in female than male mice: protective role of NO. Am J Physiol Heart Circ Physiol 284:H683–690
Cross HR, Steenbergen C, Lefkowitz RJ, Koch WJ, Murphy E (1999) Overexpression of the cardiac beta(2)-adrenergic receptor and expression of a beta-adrenergic receptor kinase-1 (betaARK1) inhibitor both increase myocardial contractility but have differential effects on susceptibility to ischemic injury. Circ Res 85:1077–1084
Cross HR, Murphy E, Steenbergen C (2002) Ca(2+) loading and adrenergic stimulation reveal male/female differences in susceptibility to ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 283:H481–489
Gabel SA, Walker VR, London RE, Steenbergen C, Korach KS, Murphy E (2005) Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. J Mol Cell Cardiol 38:289–297
Kam KW, Qi JS, Chen M, Wong TM (2004) Estrogen reduces cardiac injury and expression of beta1-adrenoceptor upon ischemic insult in the rat heart. J Pharmacol Exp Ther 309:8–15
Booth EA, Obeid NR, Lucchesi BR (2005) Activation of estrogen receptor-alpha protects the in vivo rabbit heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 289:H2039–2047
Das B, Sarkar C (2006) Similarities between ischemic preconditioning and 17beta-estradiol mediated cardiomyocyte KATP channel activation leading to cardioprotective and antiarrhythmic effects during ischemia/reperfusion in the intact rabbit heart. J Cardiovasc Pharmacol 47:277–286
Hale SL, Birnbaum Y, Kloner RA (1996) beta-Estradiol, but not alpha-estradiol, reduced myocardial necrosis in rabbits after ischemia and reperfusion. Am Heart J 132:258–262
Booth EA, Marchesi M, Kilbourne EJ, Lucchesi BR (2003) 17Beta-estradiol as a receptor-mediated cardioprotective agent. J Pharmacol Exp Ther 307:395–401
Sbarouni E, Iliodromitis EK, Bofilis E, Kyriakides ZS, Kremastinos DT (1998) Short-term estrogen reduces myocardial infarct size in oophorectomized female rabbits in a dose-dependent manner. Cardiovasc Drugs Ther 12:457–462
Sbarouni E, Iliodromitis EK, Bofilis E, Kyriakides ZS, Kremastinos DT (2003) Estrogen alone or combined with medroxyprogesterone but not raloxifene reduce myocardial infarct size. Eur J Pharmacol 467:163–168
Lee TM, Su SF, Tsai CC, Lee YT, Tsai CH (2000) Cardioprotective effects of 17 beta-estradiol produced by activation ofmitochondrial ATP-sensitive K(+)Channels in canine hearts. J Mol Cell Cardiol 32:1147–1158
Zhai P, Eurell TE, Cooke PS, Lubahn DB, Gross DR (2000) Myocardial ischemia-reperfusion injury in estrogen receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ Physiol 278:H1640–1647
Yu HP, Shimizu T, Choudhry MA, Hsieh YC, Suzuki T, Bland KI, Chaudry IH (2006) Mechanism of cardioprotection following trauma-hemorrhagic shock by a selective estrogen receptor-beta agonist: up-regulation of cardiac heat shock factor-1 and heat shock proteins. J Mol Cell Cardiol 40:185–194
Nikolic, I., Liu, D., Bell, J., Collins, J., Steenbergen, C., and Murphy, E (2007) Treatment with an estrogen receptor beta selective agonist is cardioprotective. J Molec Cellular Cardiol 42:769–780
Hsieh YC, Choudhry MA, Yu HP, Shimizu T, Yang S, Suzuki T, Chen J, Bland KI, Chaudry IH (2006) Inhibition of cardiac PGC-1alpha expression abolishes ERbeta agonist-mediated cardioprotection following trauma-hemorrhage. Faseb J 20:1109–1117
Mendelsohn ME (2000) Nongenomic, ER-mediated activation of endothelial nitric oxide synthase: how does it work? What does it mean? Circ Res 87:956–960
Nuedling S, Karas RH, Mendelsohn ME, Katzenellenbogen JA, Katzenellenbogen BS, Meyer R, Vetter H, Grohe C (2001) Activation of estrogen receptor beta is a prerequisite for estrogen-dependent upregulation of nitric oxide synthases in neonatal rat cardiac myocytes. FEBS Lett 502:103–108
Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E (2006) Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res 98:403–411
Chen J, Petranka J, Yamamura K, London RE, Steenbergen C, Murphy E (2003) Gender differences in sarcoplasmic reticulum calcium loading after isoproterenol. Am J Physiol Heart Circ Physiol 285:H2657–2662
Jones SP, Bolli R (2006) The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol 40:16–23
Costa AD, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, Critz SD (2005) Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res 97:329–336
Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS (2006) Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J 394:627–634
Martin C, Schulz R, Post H, Gres P, Heusch G (2003) Effect of NO synthase inhibition on myocardial metabolism during moderate ischemia. Am J Physiol Heart Circ Physiol 284:H2320–2324
Recchia FA, Osorio JC, Chandler MP, Xu X, Panchal AR, Lopaschuk GD, Hintze TH, Stanley WC (2002) Reduced synthesis of NO causes marked alterations in myocardial substrate metabolism in conscious dogs. Am J Physiol Endocrinol Metab 282:E197–206
Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO (2003) Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299:896–899
Otsuki M, Gao H, Dahlman-Wright K, Ohlsson C, Eguchi N, Urade Y, Gustafsson JA (2003) Specific regulation of lipocalin-type prostaglandin D synthase in mouse heart by estrogen receptor beta. Mol Endocrinol 17:1844–1855
Cappola TP, Cope L, Cernetich A, Barouch LA, Minhas K, Irizarry RA, Parmigiani G, Durrani S, Lavoie T, Hoffman EP, Ye SQ, Garcia JG, Hare JM (2003) Deficiency of different nitric oxide synthase isoforms activates divergent transcriptional programs in cardiac hypertrophy. Physiol Genomics 14:25–34
Miyazaki M, Jacobson MJ, Man WC, Cohen P, Asilmaz E, Friedman JM, Ntambi JM (2003) Identification and characterization of murine SCD4, a novel heart-specific stearoyl-CoA desaturase isoform regulated by leptin and dietary factors. J Biol Chem 278:33904–33911
Dobrzyn A, Ntambi JM (2004) The role of stearoyl-CoA desaturase in body weight regulation. Trends Cardiovasc Med 14:77–81
Lopez D, Sanchez MD, Shea-Eaton W, McLean MP (2002) Estrogen activates the high-density lipoprotein receptor gene via binding to estrogen response elements and interaction with sterol regulatory element binding protein-1A. Endocrinology 143:2155–2168
Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN (2004) Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem 279:48085–48090
Chung TH, Wang SM, Wu JC (2004) 17beta-estradiol reduces the effect of metabolic inhibition on gap junction intercellular communication in rat cardiomyocytes via the estrogen receptor. J Mol Cell Cardiol 37:1013–1022
Yu W, Dahl G, Werner R (1994) The connexin43 gene is responsive to oestrogen. Proc Biol Sci 255:125–132
Too CK, Giles A, Wilkinson M (1999) Estrogen stimulates expression of adenine nucleotide translocator ANT1 messenger RNA in female rat hearts. Mol Cell Endocrinol 150:161–167
Voss MR, Stallone JN, Li M, Cornelussen RN, Knuefermann P, Knowlton AA (2003) Gender differences in the expression of heat shock proteins: the effect of estrogen. Am J Physiol Heart Circ Physiol 285:H687–692
Hsieh YC, Yu HP, Suzuki T, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH (2006) Upregulation of mitochondrial respiratory complex IV by estrogen receptor-beta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma-hemorrhage. J Mol Cell Cardiol 41:511–521
Barros RP, Machado UF, Warner M, Gustafsson JA (2006) Muscle GLUT4 regulation by estrogen receptors ERbeta and ERalpha. Proc Natl Acad Sci USA 103:1605–1608
Pedram A, Razandi M, Aitkenhead M, Levin ER (2005) Estrogen inhibits cardiomyocyte hypertrophy in vitro. Antagonism of calcineurin-related hypertrophy through induction of MCIP1. J Biol Chem 280:26339–26348
Boengler K, Schulz R, Heusch G (2006) Connexin 43 signalling and cardioprotection. Heart 92:1724–1727
Shamaei-Tousi A, Halcox JP, Henderson B (2007) Stressing the obvious? Cell stress and cell stress proteins in cardiovascular disease. Cardiovasc Res 74:19–28
Justo R, Boada J, Frontera M, Oliver J, Bermudez J, Gianotti M (2005) Gender dimorphism in rat liver mitochondrial oxidative metabolism and biogenesis. Am J Physiol Cell Physiol 289:C372–378
Duckles SP, Krause DN, Stirone C, Procaccio V (2006) Estrogen and mitochondria: a new paradigm for vascular protection? Mol Interv 6:26–35
Stirone C, Duckles SP, Krause DN, Procaccio V (2005) Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol 68:959–965
Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO (2005) Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310:314–317
Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J (2004) Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res 95:971–980
Yan L, Ge H, Li H, Lieber SC, Natividad F, Resuello RR, Kim SJ, Akeju S, Sun A, Loo K, Peppas AP, Rossi F, Lewandowski ED, Thomas AP, Vatner SF, Vatner DE (2004) Gender-specific proteomic alterations in glycolytic and mitochondrial pathways in aging monkey hearts. J Mol Cell Cardiol 37:921–929
Pedram A, Razandi M, Wallace DC, Levin ER (2006) Functional estrogen receptors in the mitochondria of breast cancer cells. Mol Biol Cell 17:2125–2137
Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Jr Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW (2004) Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci USA 101:4130–4135
Schwend T, Gustafsson JA (2006) False positives in MALDI-TOF detection of ERbeta in mitochondria. Biochem Biophys Res Commun 343:707–711
Kanaya AM, Herrington D, Vittinghoff E, Lin F, Grady D, Bittner V, Cauley JA, Barrett-Connor E (2003) Glycemic effects of postmenopausal hormone therapy: the Heart and Estrogen/progestin Replacement Study. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 138:1–9
Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS (2000) Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA 97:12729–12734
Naaz A, Zakroczymski M, Heine P, Taylor J, Saunders P, Lubahn D, Cooke PS (2002) Effect of ovariectomy on adipose tissue of mice in the absence of estrogen receptor alpha (ERalpha): a potential role for estrogen receptor beta (ERbeta). Horm Metab Res 34:758–763
Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS (1994) Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 331:1056–1061
Lopaschuk GD (1998) Treating ischemic heart disease by pharmacologically improving cardiac energy metabolism. Am J Cardiol 82:14K–17K
Apstein CS (2000) Increased glycolytic substrate protection improves ischemic cardiac dysfunction and reduces injury. Am Heart J 139:S107–114
Lopaschuk GD, Barr R, Thomas PD, Dyck JR (2003) Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res 93:e33–37
Wahr JA, Olszanski D, Childs KF, Bolling SF (1996) Dichloroacetate enhanced myocardial functional recovery post-ischemia: ATP and NADH recovery. J Surg Res 63:220–224
Saeedi R, Wambolt RB, Parsons H, Antler C, Leong HS, Keller A, Dunaway GA, Popov KM, Allard MF (2006) Gender and post-ischemic recovery of hypertrophied rat hearts. BMC Cardiovasc Disord 6:8
Kurzelewski M, Duda M, Stanley WC, Boemke W, Beresewicz A (2004) Nitric oxide synthase inhibition and elevated endothelin increase oxygen consumption but do not affect glucose and palmitate oxidation in the isolated rat heart. J Physiol Pharmacol 55:27–38
Lei B, Matsuo K, Labinskyy V, Sharma N, Chandler MP, Ahn A, Hintze TH, Stanley WC, Recchia FA (2005) Exogenous nitric oxide reduces glucose transporters translocation and lactate production in ischemic myocardium in vivo. Proc Natl Acad Sci USA 102:6966–6971
Li J, Hu X, Selvakumar P, Russell RR, 3rd Cushman SW, Holman GD, Young LH (2004) Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab 287:E834–841
Sasaki N, Sato T, Ohler A, O’Rourke B, Marban E (2000) Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation 101:439–445
Fontaine E, Eriksson O, Ichas F, Bernardi P (1998) Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation by electron flow through the respiratory chain complex i. J Biol Chem 273:12662–12668
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Murphy, E., Steenbergen, C. Cardioprotection in females: a role for nitric oxide and altered gene expression. Heart Fail Rev 12, 293–300 (2007). https://doi.org/10.1007/s10741-007-9035-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-007-9035-0