
Abstract
Since the discovery of atrial natriuretic factor by de Bold et al., there has been tremendous progress in our understanding of the physiologic, diagnostic and therapeutic roles of the natriuretic peptides (NPs) in health and disease. Natriuretic peptides are endogenous hormones that are released by the heart in response to myocardial stretch and overload. Three mammalian NPs have been identified and characterized, including atrial natriuretic peptide (ANP or atrial natriuretic factor), B-type natriuretic peptide (BNP), and C-type natriuretic peptide (CNP). In addition, Dendroaspis natriuretic peptide (DNP) has been isolated from the venom of Dendroaspis angusticeps (the green mamba snake), and urodilatin from human urine. These peptides are structurally similar and they consist of a 17-amino-acid core ring and a cysteine bridge. Both ANP and BNP bind to natriuretic peptide receptor A (NPR-A) that are expressed in the heart and other organs. Activation of NPR-A generates an increase in cyclic guanosine monophosphate, which mediates natriuresis, inhibition of renin and aldosterone, as well as vasorelaxant, anti-fibrotic, anti-hypertrophic, and lusitropic effects. The NP system thus serves as an important compensatory mechanism against neurohumoral activation in heart failure. This provides a strong rationale for the use of exogenous NPs in the management of acutely decompensated heart failure. In this article, the therapeutic applications of NPs in the acute heart failure syndromes are reviewed. Emerging therapeutic agents and areas for future research are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
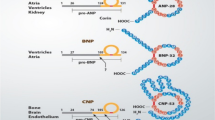
Since the discovery of atrial natriuretic factor by de Bold et al. [1], there has been tremendous progress in our understanding of the physiologic, diagnostic and therapeutic roles of the natriuretic peptides (NPs) in health and disease. Natriuretic peptides are endogenous hormones that are released by the heart in response to myocardial stretch and overload [2]. Three mammalian NPs have been identified and characterized, including atrial natriuretic peptide (ANP or atrial natriuretic factor), B-type natriuretic peptide (BNP), and C-type natriuretic peptide (CNP) [3, 4]. Moreover, Dendroaspis natriuretic peptide (DNP) was isolated from the venom of Dendroaspis angusticeps (the green mamba snake) [5] and DNP-like immunoreactivity has also been detected in the human plasma and myocardium [6]. However, the gene for DNP has yet to be identified in the human genome [7], whereas ANP, BNP, and CNP are known to be genetically distinct [3, 4]. In addition, urodilatin (URO) is an NP that has been isolated in human urine [3]. Urodilatin originates from the same common precursor as ANP, but is differentially processed [8]. All five NPs share structural similarities in a 17-amino-acid core ring and a cysteine bridge (Fig. 1) [3, 5].

In this article, the therapeutic applications of NPs in the acute heart failure syndromes (AHFS) are reviewed. Emerging therapeutic agents and areas for future research are discussed.
Rationale for the therapeutic use of natriuretic peptides in the AHFS
Both ANP and BNP are produced by myocardial cells [9]. They bind to natriuretic peptide receptor A (NPR-A) [10], which is widely distributed in the human myocardium [11]. Both NPs are released in response to myocardial wall stretch [9], which is exaggerated in heart failure (HF) where cardiac filling pressures are elevated [12, 13]. Activation of NPR-A results in an increase in cyclic guanosine monophosphate (cGMP), which mediates natriuresis, inhibition of renin and aldosterone, as well as vasorelaxant, anti-fibrotic, anti-hypertrophic, lusitropic and other effects (Fig. 2) [4, 9].

Schematic illustration of the signal transduction pathways of the natriuretic peptide/nitric oxide system and therapeutic targets for potentiation of cGMP effects [4, 7, 77]. ANP = A-type natriuretic peptide, BNP = B-type natriuretic peptide, CNP = C-type natriuretic peptide, DNP = Dendroaspis natriuretic peptide, GC = guanylate cyclase, sGC = soluble guanylate cyclase, cGMP = cyclic guanosine monophosphate, GTP = guanosine triphosphate, NEP = neutral endopeptidase, NO = nitric oxide, NPs = natriuretic peptides, NPR-A = natriuretic peptide receptor-A, NPR-B = natriuretic peptide receptor-B, NPR-C = natriuretic peptide receptor-C, PDE = phosphodiesterase
C-type natriuretic peptide was first identified in porcine brain [14]. It is of endothelial and renal cell origin with a wide distribution in the vasculature, brain, bone, epithelium, and other tissues [4, 15–17]. It is thought to act via a paracrine mechanism [18]. CNP preferentially binds to the natriuretic peptide receptor-B (NPR-B) [10, 19], which is in abundance in veins as compared with arteries [20]. Activation of NPR-B by CNP increases cGMP in vascular smooth muscle cells and mediates vasorelaxation [21]. Additional actions of CNP include reduction in cardiac preload in vivo [22]; regulation of vascular tone [21]; inhibition of vascular smooth muscle cell proliferation, hypertrophy of cardiac myocytes and growth of fibroblasts [21, 23–25]; suppression of aldosterone release [26, 27]; attenuation of myocardial ischemia-reperfusion injury (IRI) [28, 29]; and prevention of remodeling following myocardial infarction [30]. CNP might also contribute to the anti-mitogenic and vasodilatory effects of ANP and BNP, as CNP secretion can be stimulated by ANP and BNP [31]. However, it lacks significant natriuretic or diuretic effects [22, 27].
Urodilatin exhibits structural homology with the circulating human α-ANP and, in addition, possesses an extension of four amino-acids in the N-terminus (Fig. 1) [8, 32]. It also binds preferentially to NPR-A, although with less affinity as compared with that of α-ANP [33]. Urodilatin acts in the glomeruli and the inner medullary collecting ducts [34], and functions as a paracrine regulator of Na+ excretion in the kidney [8, 35].
Given these favorable cardiorenal actions, the NP system serves as an important compensatory mechanism against the neurohumoral activation in HF [9]. This provides a strong rationale for the application of exogenous NPs in the management of the AHFS.
Review of ANP, BNP, and urodilatin
Two of the NPs, ANP and BNP, are available for clinical use, whereas URO is under clinical investigation. Specifically, human recombinant ANP (carperitide) was approved in Japan in 1995 for the management of acute HF and is currently under clinical development in the United States [3, 36]. Human recombinant BNP (nesiritide) was approved in the U.S. in 2001 [37]. Urodilatin (ularitide) is currently under clinical development both in Europe and in the U.S. [3, 36].
ANP
Human ANP has been evaluated in multiple studies with a wide variation of pharmacokinetic parameters reported [38]. Generally speaking, ANP has a short half-life and a high total body clearance [38]. Nakao et al. [39] studied the pharmacokinetics of synthetic α-human ANP in six healthy male volunteers using an i.v. bolus injection (100 μg). The disappearance of α-hANP was fitted to a bi-exponential decay curve, with fast and slow half-times being 1.7 min and 13.3 min, respectively. The volume of distribution at steady state was 11.9 l and the mean plasma clearance was 1.52 l min−1 kg−1. Weidmann et al. [40] showed that in healthy men, i.v. infusion of synthetic α-hANP lowered systemic blood pressure (BP), increased glomerular filtration rate (GFR), and mediated marked natriuresis and diuresis. Hemoconcentration was observed [40]. In addition, Eiskjær and Pedersen [41] evaluated the dose-response relationship of ANP bolus injection in healthy men and reported dose-dependent increases in cGMP in plasma and urine following ANP bolus injection. The increase in cGMP correlated with the increase in urinary Na+ excretion [41]. ANP (as well as BNP and urodilatin) is cleared by NPR-C and degraded by neutral endopeptidase (NEP) 24.11 [3]. Both NPR-C and NEP are found in the kidney, vascular wall, and lung [3].
In patients with HF, Cody et al. [42] demonstrated that i.v. infusion of ANP led to natriuresis and diuresis, inhibition of renin and aldosterone, decreases in systemic BP and pulmonary capillary wedge pressure (PCWP). However, renal response was attenuated in heart failure when compared to normals [42]. This relative resistance to NP has not been well delineated, but potential explanations [3, 43] may include downregulation of NP receptors in the kidney, existence of altered forms of NPs [44], reduced production or increased degradation of cGMP; increased activity of NEP, increased renal sympathetic activity, hyperaldosteronism (increased distal tubular Na+ reabsorption), increased phosphodiesterase activity, enhanced activity of functional antagonists (such as RAAS) and reduced Na+ delivery to the distal tubules. In another study, Giles et al. [45] assessed the hemodynamic responses to human ANP in 12 patients with HF. At 30 minutes following i.v. bolus injection (4.5 μg kg−1 min−1), right atrial pressure (RAP), PCWP and heart rate (HR) decreased significantly [45]. More recently, the efficacy and safety of carperitide were evaluated in a 6-year prospective open-label registry of 3,777 patients with acute HF (51% Killip class III or IV) who were treated with a median dose of 0.085 μg kg−1 min−1of carperitide (median duration 65 h). It was reported that 82% of patients improved clinically [46].
BNP
Nesiritide, a purified preparation of human BNP, is manufactured from Escherichia coli using recombinant DNA technology [47]. Recombinant BNP exhibits similar physiologic actions as endogenous BNP [47, 48]. The distribution half-life and the mean terminal elimination half-life of nesiritide are approximately 2 min and 18 min, respectively [47]. The time to steady state level is less than 90 min and the mean volume of distribution (at steady state) is 0.19 l kg−1 [47]. Clearance of nesiritide is achieved through 3 mechanisms: binding to the NPR-C on cell surface, degradation by neutral endopeptidase, and renal filtration [47]. Dosage reduction is not needed in patients with renal insufficiency [47, 48].
Nesiritide has been shown to lower filling pressure, decrease systemic and pulmonary vascular resistance and increase cardiac output in a dose-dependent manner [49]. At 0.01 μg kg−1 (the currently recommended dose), nesiritide significantly lowers LV filling pressure [49]. The effects of nesiritide on GFR and renal blood flow (RBF) are variable [49].
Intravenous nesiritide has been studied in a number of randomized clinical trials [50]. Colucci et al. [51] evaluated the efficacy of nesiritide in the treatment of decompensated HF. Patients who were hospitalized for symptomatic HF were recruited into either the efficacy trial of double-blind design or the open-label comparative trial. In the efficacy trial (which required placement of a pulmonary arterial catheter), 127 patients who had PCWP ≥ 18 mmHg and a cardiac index ≤2.7 l min−1 m−2 were randomized to placebo or nesiritide (0.015 μg kg−1 min−1 or 0.03 μg kg−1 min−1 infusion) for 6 h [51]. Nesiritide decreased PCWP by 6 mmHg and 9.6 mmHg, respectively (vs. an increase of 2 mmHg with placebo) and was associated with significant improvement in dyspnea, fatigue, and global clinical status [51]. In the comparative trial, 305 patients were randomized to nesiritide (0.015 μg kg−1 min−1 or 0.03 μg kg−1 min−1) or standard therapy. The improvements in dyspnea, fatigue, and overall clinical status in nesiritide-treated patients were sustained for up to 7 days and were similar to those who received standard therapy [51].
The Vasodilation in the Management of Acute CHF (VMAC) study was a randomized, double-blind trial to compare the efficacy and safety of i.v. nesiritide, i.v. nitroglycerin (NTG), and placebo in 489 patients with decompensated heart failure and dyspnea at rest (246 patients had placement of pulmonary arterial catheter) [52]. Nesiritide, NTG, or placebo was given for 3 h, followed by nesiritide or NTG for 24 h. It was demonstrated that nesiritide reduced mean PCWP significantly more than either NTG or placebo at 3 h and significantly more than NTG at 24 h. There was also significant improvement in dyspnea at 3 h over placebo, but no difference was detected between nesiritide-treated and NTG-treated patients.
Additional issues on nesiritide are discussed under ‘Safety’ in this review. The interested reader is referred to recent clinical practice guidelines for further information [53–55].
Urodilatin
Urodilatin has been evaluated in a number of studies. Carstens et al. [32] studied the pharmacokinetics and renal pharmacodynamics of URO in 12 healthy men in a randomized, double-blind, crossover study. Urodilatin was administered as a 90-min infusion. The kinetics of URO was characterized by a large apparent volume of distribution (43.7 l), a high total body clearance (5.358 l min−1), and a short plasma half-life of 5.57 min [32]. Of the infused URO, less than 1% was recovered in urine. In these healthy men, URO exerted natriuretic and diuretic effects with lowering of mean blood pressure [32]. Dorner et al. [56] conducted a randomized, double-blind, placebo-controlled, dose-finding study to evaluate the hemodynamic effects of continuous infusion of URO (7.5, 15, and 22.5 ng kg−1 min−1) in healthy males. It was found that URO 7.5 ng kg−1 min−1 and 15 ng kg−1 min−1 were well tolerated and that URO at 15 ng kg−1 min−1 increased urine flow, filtration fraction, renal and systemic vascular resistance without change in GFR or blood pressure [56]. However, at 22.5 ng kg−1 min−1, systemic hypotension, nausea and dizziness occurred, requiring termination of infusion [56]. In addition, Bestle et al. [57] evaluated the cardiovascular, endocrine, and renal effects of i.v. infusion of URO in a randomized, double-blind study. Urodilatin (5, 10, 20, 40 ng kg−1 min−1) or placebo was administered over 2 h to eight healthy men (on five separate occasions for each subject). Both plasma and urinary cGMP increased following urodilatin infusion [57]. Urodilatin 5 ng kg−1 min−1 and 10 ng kg−1 min−1 did not change mean arterial pressure (MAP) or HR, but decreased stroke volume (10% and 20%, respectively) and cardiac output (7% and 16%). In contrast, URO 20 and 40 ng kg−1 min−1 decreased MAP (6% and 14%) and stroke volume (17% and 21%), and increased HR (15% and 38%) with cardiac output remained unchanged. Importantly, the renin-angiotensin-aldosterone system (RAAS) was found to be suppressed by URO at 5, 10, and 20 ng kg−1 min−1 but was activated by URO 40 ng kg−1 min−1, the latter was attributed to a decrease in renal perfusion pressure and increased activity of renal sympathetic nerves [57].
Urodilatin has been compared with other NPs. Saxenhofer et al. [58] evaluated an i.v. bolus injection of URO (25, 50, 100 μg) as compared with ANP (50 μg) or placebo. URO at 100 μg lowered diastolic BP, whereas ANP decreased both systolic and diastolic BP. A dose-dependent increase in HR was detected [58]. In addition, dose-dependent increases in urinary cGMP and Na+ excretion were observed, with URO being more potent than ANP [58]. There was an increase in GFR following administration of URO 50 μg and 100 μg. There were no changes in effective RBF, plasma renin, aldosterone, or catecholamines [58].
In a randomized, double-blind, ascending-dose, placebo-controlled safety trial, Mitrovic et al. [59] evaluated the effects of a 24-h i.v. infusion of URO in the treatment of decompensated chronic HF in 24 patients. The patients were randomized to URO 7.5, 15 and 30 ng kg−1 min−1 (in ascending order) or placebo. It was found that URO (15 ng kg−1 min−1 and 30 ng kg−1 min−1) significantly reduced PCWP and RAP, as well as N-terminal pro-BNP, as compared with baseline [59]. Despite a lack of significant difference in 24-h urine output among the 4 groups, it was noted that more patients in the placebo group and the low-dose URO group had received loop diuretics.
The SIRIUS-II (Safety and efficacy of an Intravenous placebo controlled Randomized Infusion of Ularitide in a prospective double-blind Study in patients with symptomatic decompensated chronic heart failure) trial evaluated URO in 221 HF patients who had dyspnea at rest or with minimal exertion [60]. Patients (mean age 61 years, 78% men, 72% LVEF < 30%) were randomized to placebo or URO infusion at 7.5, 15, or 30 ng kg−1 min−1 for 24 h. Preliminary results revealed that about 40% of patients in each of the URO-treated groups showed improvement, versus approximately 25% in the placebo group [60]. The decrease in PCWP was 11 mmHg in patients treated with URO 30 ng kg−1 min−1 vs. 4 mmHg in the placebo group. Mean systolic BP decreased by up to 15 mmHg without change in HR (URO 15 ng kg−1 min−1 or 30 ng kg−1 min−1) [60]. No additional diuresis was observed in URO-treated patients. Serum creatinine increased to a similar extent in the placebo group and two of the URO-treated groups (7.5 and 30 ng kg−1 min−1). Death occurred in 7 placebo-treated and 5 URO-treated patients [60].
Novel routes of administration
Alternative route of peptide administration remains to be an important area of investigation. Recent reports of intra-renal administration of BNP in experimental [61] and clinical [62, 63] studies appear promising. For example, Heywood et al. [63] evaluated intra-renal infusion of nesiritide in cardiac transplant patients and reported increases in both GFR and RBF. Importantly, the greatest renal benefit was observed when hypotension was minimized [63], a finding that has also been reported in another patient population with AHFS [64]. Emerging studies on peri-operative administration of NPs are also in support of a conservative dosing regimen [65–67]. Indeed, previous canine [68] and human data [3, 57, 63, 64, 69, 70] appear to be consistent with the notion that hypotension-induced activation of the RAAS and of the sympathetic nervous system may act as an underlying mechanism for the development of renal dysfunction (Fig. 3). Whether avoidance of systemic hypotension (with judicious dosing or with intra-renal administration) would extend the application of NPs to the critically ill cardiac patient remains to be explored in experimental studies.

The subcutaneous route continues to be an attractive option for the delivery of BNP, as shown in previous experimental [71–74] and clinical studies [75]. In one canine study [73], tolerance was not observed following chronic administration of BNP. Further studies are needed in this area, as interactions between the NO and the NP systems have been observed [21, 76] and tolerance is a recognized phenomenon with the former [77].
Development of orally available peptides has long been a challenge, given various barriers to protein absorption and penetration [78]. Recently, Cataliotti et al. [79] reported the application of proprietary technology which enabled oral delivery of BNP by covalently attaching short, amphiphilic oligomers to peptides. In normal conscious dogs, this novel oral conjugated human BNP activated cGMP and exerted hypotensive effects [79]. Recently, Cataliotti et al. [80] further demonstrated in a canine model of acute hypertension that a more advanced oral conjugated human BNP significantly increased cGMP and lowered MAP. The availability of an orally active NP such as BNP could lead to a much wider application of these peptides with such favorable actions as discussed above including their use in human hypertension for both cardiovascular and renal protection. Here clinical trials will be most important to define the potential safety and efficacy of oral NP in the setting of such an important disease such as hypertension.
Wang et al. [81] reported the synthesis of a long-acting fusion hormone from recombinant BNP and human serum albumin, AlbuBNP, which exhibited an extended elimination half-life of 12–19 h in mice [81]. The effective concentration for 50% response (EC50) in NPR-A/cGMP assay was 28.4 nM and 0.46 nM for AlbuBNP and BNP, respectively [81].
The relevance of chronic use of BNP to the AHFS is that this strategy not only provides opportunities of stabilization during the clinical course of HF, but also might partially circumvent limitations associated with delayed drug administration for acute HF and/or ischemia. Evaluation of its use as an oral agent in hypertension should also be a top priority.
Safety
In a prospective registry of 3,777 patients with acute HF treated with carperitide over a 6-year period, Suwa et al. [46] reported a 17% incidence of adverse events, with the most common one being hypotension (during the first 3 h of carperitide infusion), which resolved spontaneously in 96% of patients. The median dose was 0.085 μg kg−1 min−1. Clinical improvement was noted in 82% of the patients [46].
Recent analyses [82–84] of clinical trial data on nesiritide have raised concerns about adverse renal effects and increase in mortality. An expert panel was convened to provide guidance amidst these concerns [85]. It was recommended that the use of nesiritide to be limited to patients with acutely decompensated HF who have dyspnea at rest and that nesiritide not be used in place of diuretics, given the lack of sufficient evidence [85]. Moreover, further clinical studies were recommended to evaluate the effect of nesiritide on survival [85]. The use of nesiritide at the dosages described in the package insert (0.01–0.03 μg·kg−1 min−1) was noted to be associated with a dose-dependent increase in serum creatinine [85]. The mechanism for this untoward effect has not been well delineated, although it is likely multifactorial [3], including NP-induced systemic hypotension, with subsequent reduction in renal perfusion pressure and activation of the RAAS (Fig. 3). Indeed, excess hypotension is a known adverse effect of nesiritide. In the VMAC trial [86], the incidence of symptomatic hypotension was 4% and the mean duration of hypotension was 2.2 h. Most episodes resolved spontaneous or in response to i.v. fluid administration. Of note, concomitant administration of oral angiotensin converting enzyme inhibitors may increase the risk of symptomatic hypotension [46, 47]. Systemic hypotension has been reported with the use of ANP [87] and URO [56, 57, 88]. Hypotension, nausea, and dizziness were experienced by healthy subjects during URO infusion at 22.5 ng kg−1 min−1, requiring cessation of infusion [56]. In another study of URO [57], 3 healthy subjects (1 received 20 ng kg−1 min−1 and 2 received 40 ng kg−1 min−1) had syncope, which resolved with cessation of infusion and assumption of Trendelenburg position.
Newer cyclic GMP activating drugs for heart failure
In acute and chronic HF syndromes, pathways involving cGMP may be disrupted [77, 89]. One potential mechanism is the reduced availability of nitric oxide (NO), as in impaired production or increased degradation [77]. Other mechanisms may include inadequate release of ligands (NPs) for particulate guanylate cyclase (pGC) [77, 90], or release of altered forms [44]. Newer cGMP activating drugs for HF may act through the pGC pathway or the soluble guanylate cyclase (sGC) pathway, which are exemplified by exogenous NPs and the sGC stimulators/activators, respectively (Fig. 2).
Dendroaspis natriuretic peptide and other snake venoms
In 1992, Schweitz et al. [5] identified a new member of NP family in the venom of Dendroaspis angusticeps (the green mamba snake). This 38-amino-acid peptide, named Dendroaspis natriuretic peptide (DNP), shares the core ring conformation with other NPs and, in addition, has a unique 15-amino-acid extension in the C terminus (Fig. 1) [5].
DNP exhibits functional features that are characteristic of NPs. In pre-contracted rat aortic strips, DNP (100 nM) induced relaxation to a similar extent as ANP [5]. In cultured rat aortic myocytes and bovine aortic endothelial cells, DNP induced concentration-dependent increases in cGMP. Moreover, in binding experiments (where cultured rat aortic myocytes were exposed to 125I-ANP in the presence of increasing concentrations of DNP or unlabeled ANP), DNP prevented 125I-ANP from binding to cultured rat aortic myocytes [5]. More recently, in the human myocardium, Singh et al. [11] demonstrated the selectivity of DNP for NPR-A using a radioiodinated analog of DNP. In competition binding experiments, the rank order of potency for NPR-A was DNP > BNP–ANP \( \gg \) CNP [11]. DNP-like immunoactivity has been detected in isolated arteries and veins from humans [91], in human plasma (with elevated levels in HF patients versus normal subjects) and atrial myocardium [6]. DNP has been shown to exert vasorelaxant effects on canine coronary arteries (through pGC activation) [92] and on isolated human arteries and veins (with more prominent vasorelaxation in arteries) [91]. Most recently, Singh et al. [93] reported saturable and subnanomolar-affinity binding of [125I]-DNP to NPR-A in human mammary artery, with localization to the vascular smooth muscle layer using confocal microscopy and fluorescent dual-labeling immunocytochemistry. Moreover, DNP completely reversed endothelin-1-induced vasoconstriction with a nanomolar range of potency [93].
In vivo cardiovascular actions of synthetic DNP were evaluated in a canine study by Lainchbury et al. [94]. In anesthetized dogs, DNP infusion (10 ng kg−1 min−1 for 30 min then 50 ng kg−1 min−1 for 30 min) significantly reduced MAP and PCWP vs. vehicle [94]. Increases in DNP-like immunoreactivity and cGMP were detected in plasma. In conscious dogs, DNP reduced LV preload and afterload, slightly enhanced contractility, and improved relaxation [94].
The renal actions of i.v. infusion of synthetic DNP were assessed by Lisy et al. [95]. In normal dogs, DNP-like immunoreactivity was detected in plasma and urine, as well as in the atrial myocardium [95]. Following DNP infusion (10 ng kg−1 min−1 and 50 ng kg−1 min−1), natriuretic and diuretic effects were observed, together with increases in both plasma and urinary cGMP [95]. Moreover, a decrease in distal tubular reabsorption of Na+ was detected without change in GFR or RBF [95]. These beneficial cardiorenal actions of DNP were also observed in dogs with pacing-induced HF, with further demonstrations of suppressed plasma renin activity, increased GFR (with natriuresis and diuresis), and reduced cardiac filling pressures [96]. In another study [97], intra-renal infusion of DNP (5 ng kg−1 min−1) into normal anesthetized dogs resulted in significant increases in plasma and urinary cGMP, RBF and GFR, and Na+ excretion, along with a significant decrease in distal fraction Na+ reabsorption [97]. NEP inhibition did not augment the renal actions, suggesting that DNP might be resistant to degradation by NEP [97].
Other NPs have been identified from snake venoms, which are a valuable source of peptides for drug discovery and development [98–100]. Bazaa et al. [101] performed a comparative proteomic analysis of venoms of the three most important vipers of Tunisia: Cerastes cerastes, Cerastes vipera, and Macrovipera lebetina, and reported the presence of NPs in the venom of M. lebetina. Two of the NPs were found to be identical in sequence and mass to previously reported polypeptides isolated from M. lebetina [101] and they exhibited anti-platelet activities (inhibition of human platelet aggregation by 50% at 100 nM) [102]. Notably, antiplatelet activities have also been reported in a 37-residue peptide, Pseudocerastes persicus natriuretic peptide (PNP), which was isolated from the venom of the Iranian viper, Pseudocerastes persicus [103]. In addition, Michel et al [104] reported the isolation of two N-terminally truncated forms of CNP from the venom of Trimeresurus flavoviridis (habu snake): Tf-CNP(6-22) and Tf-CNP(3-22) [104]. The former exhibited concentration-dependent vasorelaxation in rat aortic strips (potency about 45 times lower than that of human ANP) and a weak diuretic effect [104].
Novel NPs have also been isolated from Oxyuranus microlepidotus (the inland taipan),[105] and Micrurus corallinus (the South American coral snake) [106]. These NPs were noted to have long C-terminal extensions containing proline residues [105, 106], which are features shared by DNP [5]. Among the 3 NPs (TNP-a, TNP-b, and TNP-c) isolated from the inland taipan, TNP-c was of similar potency to ANP and DNP, resulting in near complete relaxation in pre-contracted aortae [105]. Recently, St. Pierre et al. [107, 108] also reported comprehensive studies on the identification and comparative analysis of venom-gland-specific genes from Oxyuranus scutellatus (the costal taipan) and other snake species. Notably, from Pseudonaja textiles (the common brown snake), 2 isoforms (PtNP-a and PaNP-c) of an NP were found to inhibit angiotensin converting enzyme (ACE) in a dose-dependent manner [108]. One of these isoforms, PtNP-a, also exhibited cGMP-stimulating property. In addition, there have been reports of identification of bradykinin-potentiating peptides and CNP from snake venom [109–112]. Conceivably, the concomitant presence of an ACE-inhibiting property in an NP is an attractive attribute for novel heart failure therapies, especially as an oral agent for chronic administration. Whether these observations can be translated into clinical application await further studies.
Chimeric natriuretic peptides
Over the past 15 years, our research group has been actively involved in advancing the concept of chimeric NPs as novel designer peptides with unique pharmacological profile for the treatment of HF [113]. Vasonatrin peptide (VNP), a chimera that was synthesized based on the 22-amino-acid structure of CNP and the 5-amino-acid C-terminus of ANP, exerts venodilating and natriuretic effects that are characteristic of CNP and ANP, respectively [113]. Notably, VNP exhibits more potent vasorelaxant actions in both arteries and veins, as compared with ANP and CNP, and also possesses a unique arterial dilating effect that was found in neither of the parent peptides [113]. Thus, chimeric NPs may afford the opportunity of tailoring peptide design according to the underlying cardiorenal pathophysiology and the known structural requirements for specific pharmacological actions. More recently, another chimeric NP, CD-NP, was synthesized combining the ring-structure of CNP and the 15-amino-acid linear N-terminus of DNP [114]. Lisy et al. [115] observed in normal anesthetized dogs that CD-NP activates cGMP, decreases cardiac filling pressures, induces natriuresis and diuresis, and inhibits renin release. Additional chimeric NPs are in various stages of discovery in our laboratory.
Combination therapy
B-type natriuretic peptide has been tested in combination with furosemide, a loop diuretic, in dogs with pacing-induced HF by Cataliotti et al. [116], who reported augmentation of natriuresis and diuresis, improvement of GFR and attenuation of aldosterone release, as compared with furosemide alone. In another canine study of pacing-induced HF, Chen et al. [72] evaluated subcutaneous BNP in combination with omapatrilat, a vasopeptidase inhibitor, and showed greater improvement in both cardiac output and filling pressures, as well as augmentation of natriuresis and GFR, than either agent alone. More recently, co-administration of sildenafil, a type-V phosphodiesterase inhibitor, and BNP was shown to enhance renal cGMP and improve renal function in dogs with pacing-induced overt HF [74]. In addition, BNP has been tested in combination with BAY 58-2667, a direct nitric oxide-independent sGC activator, [117] and tolvaptan, a vasopressin V2 receptor antagonist, [118] with promising results.
Soluble guanylyl cyclase (sGC) stimulators and activators
Soluble guanylyl cyclase, the principal intracellular receptor for NO, is a heterodimer consisting of an α- and a heme-containing β-subunit [119–121]. Activation of sGC by NO results in a marked elevation in cGMP, which interacts with effector molecules and plays major physiological roles, such as in platelet aggregation, vasodilation, and neurotransmission [119, 120, 122].
A number of sGC stimulators and activators have been synthesized [77]. In 1994, a novel benzylindazole compound, YC-1, was reported to be a direct sGC activator in rabbit platelets, resulting in concentration-dependent inhibition of agonist-induced platelet aggregation and an increase in cGMP [123]. It also inhibited agonist-induced human platelet aggregation in a concentration-dependent manner [124] and exhibited other pharmacologic effects (see [77] for review). In 2001, Stasch et al. [125] reported the identification of BAY 41-2272, a pyrazolopyridine and a direct stimulator of sGC, and a regulatory site on sGC in the α1 subunit. It was shown that BAY 41-2272 stimulated this site (0.1–100 μM) via an NO-independent but heme-dependent mechanism. In pre-constricted rabbit aortic rings, BAY 41-2272 induced relaxation in a concentration-dependent manner (50% inhibitory concentration, or IC50, 304 nM) [125]. It inhibited collagen-induced aggregation of human platelets in vitro (IC50 36 nM) and significantly prolonged rat tail-bleeding time in vivo (0.3–3.0 mg kg−1 p.o.) [125]. In spontaneously hypertensive rats, it elicited a dose-dependent decrease in mean BP (1–10 mg kg−1 p.o.) [125]. Moreover, in a high-renin, low-NO rat model of hypertension, BAY 41-2722 (10 mg kg−1 p.o.) abolished the increase in systolic BP induced by the NO synthase inhibitor, l-NAME (N-nitro-l-arginine methylester), and significantly reduced mortality as compared with control [125]. Notably, tolerance was not detected [125].
The potential of BAY 41-2272 as a novel therapy in experimental HF was evaluated by Boerrigter et al. [126]. In a canine model of pacing-induced severe HF, BAY 41-2272 (10 μg kg−1 min−1) significantly reduced MAP, pulmonary arterial pressure, PCWP, and systemic vascular resistance (the latter was decreased at 2 μg kg−1 min−1) [126]. BAY 41-2272 (10 μg kg−1 min−1) significantly increased cardiac output and RBF from baseline [126]. There were no changes in GFR, urinary Na+, or the hormonal profile of the RAAS [126]. When BAY 41-2272 was compared with NTG (1 μg kg−1 min−1 and 5 μg kg−1 min−1), similar hemodynamic findings were observed, with the exceptions of RAP and pulmonary vascular resistance, which were significantly reduced only by NTG. Taken together, these findings suggest that BAY-41-2272 primarily acts as a pure arterial vasodilator with salutary cardiorenal actions without activating RAAS.
More recently, BAY 58-2667, an amino dicarboxylic acid and NO-independent sGC activator with vasorelaxant and anti-aggregatory effects [122, 127], and BNP, a pGC stimulator, were evaluated in combination and alone by Boerrigter et al. [117]. Concomitant activation of sGC and pGC was shown to be beneficial in augmenting cardiorenal actions in a canine model of pacing-induced HF [117]. BAY 58-2667 is currently being evaluated in clinical studies [77].
Future research and clinical applications
Based on the foregoing discussion, additional studies on nesiritide focusing on any potential adverse effects on mortality and renal function in patients with ADHF are urgently needed. It is anticipated that future clinical trials, including the ETNA (Evaluating Treatment with Nesiritide in Acute Decompensated Heart Failure) trial [128] and the ASCEND-HF (Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure) trial [129], would provide important information on these issues.
Emerging indications for NPS beyond heart failure
Multiple studies support the notion that NPs are protective against myocardial ischemia-reperfusion injury [130]. These cardioprotective properties may be particularly relevant in the management of patients with AHFS and concomitant acute coronary syndromes (ACS), as coronary artery disease (CAD) is common among patients with HF and ACS has recently been shown to be the most important precipitating factor of new-onset AHF in the EuroHeart Survey II [131]. Future clinical trials are needed to test the hypothesis that NPs are cardioprotective in the setting of myocardial ischemia.
Recent perioperative studies in patients undergoing coronary artery bypass graft surgery [65, 66, 132] and cardiac transplantation [67] have documented salutary renal effects of low-dose NPs (such as nesiritide 0.01 μg kg−1 min−1 without bolus [65]). Additional studies are needed to confirm these encouraging results. Nonetheless, these observations of enhanced renal function in response to NPs such as nesiritide and ANP reinforce their inherent renoprotective properties at least at low doses. Whether this strategy would be applicable to patients with pre-existing ventricular dysfunction and/or CAD undergoing high-risk non-cardiac surgery deserves evaluation. The bronchodilating effects of NPs [133] may confer additional benefits.
Conclusion
The AHFS continues to be a challenging public health problem. Despite significant advances in our knowledge in the field of NPs, much work remains to be done in refining current treatment strategies, in minimizing side effects, and in identifying opportunities in drug discovery.
References
de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H (1981) A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci 28:89–94
Boerrigter G, Burnett JC Jr (2004) Cardiorenal syndrome in decompensated heart failure: prognostic and therapeutic implications. Curr Heart Fail Rep 1:113–120
Chen HH, Burnett JC Jr (2006) Clinical application of the natriuretic peptides in heart failure. Eur Heart J 8(Suppl E):E18–E25
Potter LR, Abbey-Hosch S, Dickey DM (2006) Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev 27:47–72
Schweitz H, Vigne P, Moinier D, Frelin C, Lazdunski M (1992) A new member of the natriuretic peptide family is present in the venom of the green mamba (Dendroaspis angusticeps). J Biol Chem 267:13928–13932
Schirger JA, Heublein DM, Chen HH, Lisy O, Jougasaki M, Wennberg PW et al (1999) Presence of Dendroaspis natriuretic peptide-like immunoreactivity in human plasma and its increase during human heart failure. Mayo Clin Proc 74:126–130
Margulies KB, Burnett JC Jr (2006) Visualizing the basis for paracrine natriuretic peptide signaling in human heart. Circ Res 99:113–115
Forssmann W, Meyer M, Forssmann K (2001) The renal urodilatin system: clinical implications. Cardiovasc Res 51:450–462
Burnett JC Jr, Costello-Boerrigter L, Boerrigter G (2004) Alterations in the kidney in heart failure: the cardiorenal axis in the regulation of sodium homeostasis. In: Mann DL (ed) Heart failure: a companion to Braunwald’s heart disease. Elsevier Inc, Philadelphia, pp 279–289
Suga S, Nakao K, Hosoda K, Mukoyama M, Ogawa Y, Shirakami G et al (1992) Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinology 130:229–239
Singh G, Kuc RE, Maguire JJ, Fidock M, Davenport AP (2006) Novel snake venom ligand Dendroaspis natriuretic peptide is selective for natriuretic peptide receptor-A in human heart: downregulation of natriuretic peptide receptor-A in heart failure. Circ Res 99:183–190
Burnett JC Jr, Kao PC, Hu DC, Heser DW, Heublein D, Granger JP et al (1986) Atrial natriuretic peptide elevation in congestive heart failure in the human. Science 231:1145–1147
Mukoyama M, Nakao K, Hosoda K, Suga S, Saito Y, Ogawa Y et al (1991) Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J Clin Invest 87:1402–1412
Sudoh T, Minamino N, Kangawa K, Matsuo H (1990) C-type natriuretic peptide (CNP): a new member of natriuretic peptide family identified in porcine brain. Biochem Biophys Res Commun 168:863–870
Stingo AJ, Clavell AL, Heublein DM, Wei CM, Pittelkow MR, Burnett JC Jr (1992) Presence of C-type natriuretic peptide in cultured human endothelial cells and plasma. Am J Physiol 263:H1318–1321
Del Ry S, Passino C, Emdin M, Giannessi D (2006) C-type natriuretic peptide and heart failure. Pharmacol Res 54:326–333
Garbers DL, Chrisman TD, Wiegn P, Katafuchi T, Albanesi JP, Bielinski V et al (2006) Membrane guanylyl cyclase receptors: an update. Trends Endocrinol Metab 17:251–258
Ahluwalia A, MacAllister RJ, Hobbs AJ (2004) Vascular actions of natriuretic peptides. Cyclic GMP-dependent and -independent mechanisms. Basic Res Cardiol 99:83–89
Koller KJ, Lowe DG, Bennett GL, Minamino N, Kangawa K, Matsuo H et al (1991) Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide (CNP). Science 252:120–123
Wei CM, Aarhus LL, Miller VM, Burnett JC Jr (1993) Action of C-type natriuretic peptide in isolated canine arteries and veins. Am J Physiol 264:H71–H73
Wennberg PW, Miller VM, Rabelink T, Burnett JC Jr (1999) Further attenuation of endothelium-dependent relaxation imparted by natriuretic peptide receptor antagonism. Am J Physiol 277:H1618–H1621
Stingo AJ, Clavell AL, Aarhus LL, Burnett JC Jr (1992) Cardiovascular and renal actions of C-type natriuretic peptide. Am J Physiol 262:H308–H312
Furuya M, Yoshida M, Hayashi Y, Ohnuma N, Minamino N, Kangawa K et al (1991) C-type natriuretic peptide is a growth inhibitor of rat vascular smooth muscle cells. Biochem Biophys Res Commun 177:927–931
Cao L, Gardner DG (1995) Natriuretic peptides inhibit DNA synthesis in cardiac fibroblasts. Hypertension 25:227–234
Tokudome T, Horio T, Soeki T, Mori K, Kishimoto I, Suga S-i et al (2004) Inhibitory effect of C-type natriuretic peptide (CNP) on cultured cardiac myocyte hypertrophy: interference between CNP and endothelin-1 signaling pathways. Endocrinology 145:2131–2140
Igaki T, Itoh H, Suga SI, Hama N, Ogawa Y, Komatsu Y et al (1998) Effects of intravenously administered C-type natriuretic peptide in humans: comparison with atrial natriuretic peptide. Hypertens Res 21:7–13
Hunt PJ, Richards AM, Espiner EA, Nicholls MG, Yandle TG (1994) Bioactivity and metabolism of C-type natriuretic peptide in normal man. J Clin Endocrinol Metab 78:1428–1435
Hobbs A, Foster P, Prescott C, Scotland R, Ahluwalia A (2004) Natriuretic peptide receptor-C regulates coronary blood flow and prevents myocardial ischemia/reperfusion injury: novel cardioprotective role for endothelium-derived C-type natriuretic peptide. Circulation 110:1231–1235
Scotland RS, Cohen M, Foster P, Lovell M, Mathur A, Ahluwalia A et al (2005) C-type natriuretic peptide inhibits leukocyte recruitment and platelet-leukocyte interactions via suppression of P-selectin expression. Proc Natl Acad Sci USA 102:14452–14457
Soeki T, Kishimoto I, Okumura H, Tokudome T, Horio T, Mori K et al (2005) C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. J Am Coll Cardiol 45:608–616
Nazario B, Hu RM, Pedram A, Prins B, Levin ER (1995) Atrial and brain natriuretic peptides stimulate the production and secretion of C-type natriuretic peptide from bovine aortic endothelial cells. J Clin Invest 95:1151–1157
Carstens J, Jensen KT, Pedersen EB (1998) Metabolism and action of urodilatin infusion in healthy volunteers. Clin Pharmacol Ther 64:73–86
Saxenhofer H, Fitzgibbon WR, Paul RV (1993) Urodilatin: binding properties and stimulation of cGMP generation in rat kidney cells. Am J Physiol 264:F267–273
Koike J, Nonoguchi H, Terada Y, Tomita K, Marumo F (1993) Effect of urodilatin on cGMP accumulation in the kidney. J Am Soc Nephrol 3:1705–1709
Goetz K, Drummer C, Zhu JL, Leadley R, Fiedler F, Gerzer R (1990) Evidence that urodilatin, rather than ANP, regulates renal sodium excretion. J Am Soc Nephrol 1:867–874
Gheorghiade M, Zannad F, Sopko G, Klein L, Pina IL, Konstam MA et al (2005) Acute heart failure syndromes: Current state and framework for future research. Circulation 112:3958–3968
Burnett JC (2005) Nesiritide: new hope for acute heart failure syndromes? Eur Heart J 7(Suppl B):B25–B30
Tan AC, Russel FG, Thien T, Benraad TJ (1993) Atrial natriuretic peptide. An overview of clinical pharmacology and pharmacokinetics. Clin Pharmacokinet 24:28–45
Nakao K, Sugawara A, Morii N, Sakamoto M, Yamada T, Itoh H et al (1986) The pharmacokinetics of alpha-human atrial natriuretic polypeptide in healthy subjects. Eur J Clin Pharmacol 31:101–103
Weidmann P, Hasler L, Gnadinger MP, Lang RE, Uehlinger DE, Shaw S et al (1986) Blood levels and renal effects of atrial natriuretic peptide in normal man. J Clin Invest 77:734–742
Eiskjaer H, Pedersen EB (1993) Dose-response study of atrial natriuretic peptide bolus injection in healthy man. Eur J Clin Invest 23:37–45
Cody RJ, Atlas SA, Laragh JH, Kubo SH, Covit AB, Ryman KS et al (1986) Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J Clin Invest 78:1362–1374
Lindenfeld J, Schrier RW (2007) The kidney in heart failure. In: Hosenpud JD, Greenberg BH (eds) Congestive heart failure, 3rd edn.Lippincott Williams & Wilkins, Philadelphia, PA, pp 243–260
Hawkridge AM, Heublein DM, Bergen HR 3rd, Cataliotti A, Burnett JC Jr, Muddiman DC (2005) Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc Natl Acad Sci USA 102:17442–17447
Giles TD, Quiroz AC, Roffidal LE, Marder H, Sander GE (1991) Prolonged hemodynamic benefits from a high-dose bolus injection of human atrial natriuretic factor in congestive heart failure. Clin Pharmacol Ther 50:557–563
Suwa M, Seino Y, Nomachi Y, Matsuki S, Funahashi K (2005) Multicenter prospective investigation on efficacy and safety of carperitide for acute heart failure in the ‘real world’ of therapy. Circ J Mar 69(3):283–90
Keating GM, Goa KL (2003) Nesiritide: a review of its use in acute decompensated heart failure. Drugs 63:47–70
Sackner-Bernstein JD, Skopicki H, Aaronson KD (2006) Natriuretic peptides for the treatment of heart failure. In: Feldman A (ed) Heart failure: pharmacologic management. Blackwell Publishing, Malden, MA, pp 154–171
Heart Failure Society of America. HFSA (2006) Comprehensive heart failure practice guideline: Section 12: Evaluation and management of patients with acute decompensated heart failure. J Card Fail 12:e86–e103
Emdin M, Clerico A (2006) Cardiac natriuretic hormone system as target for cardiovascular therapy. In: Clerico A, Emdin M (eds) Natriuretic peptides: the hormones of the heart. Springer-Verlag, New York, pp 161–175
Colucci WS, Elkayam U, Horton DP, Abraham WT, Bourge RC, Johnson AD et al (2000) for the nesiritide study group. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure N Engl J Med 343:246–253
Publication Committee for the VMAC Investigators (Vasodilation in the Management of Acute CHF) (2002) Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 287:1531–1540 [Erratum, JAMA (2002) 1288:1577]
Hunt SA (2005) ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to update the 2001 guidelines for the evaluation and management of heart failure). J Am Coll Cardiol 46:e1–e82
Nieminen MS, Bohm M, Cowie MR, Drexler H, Filippatos GS, Jondeau G et al (2005) Executive summary of the guidelines on the diagnosis and treatment of acute heart failure: the task force on acute heart failure of the European society of cardiology. Eur Heart J 26:384–416
Adams KF, Lindenfeld J, Arnold JMO, Baker DW, Barnard DH, Baughman KL et al HFSA (2006) Comprehensive Heart Failure Practice Guidelines. J Cardiac Failure 12:e1–e122
Dorner GT, Selenko N, Kral T, Schmetterer L, Eichler HG, Wolzt M (1998) Hemodynamic effects of continuous urodilatin infusion: a dose-finding study. Clin Pharmacol Ther 64:322–330
Bestle MH, Olsen NV, Christensen P, Jensen BV, Bie P (1999) Cardiovascular, endocrine, and renal effects of urodilatin in normal humans. Am J Physiol 276:R684–R695
Saxenhofer H, Raselli A, Weidmann P, Forssmann WG, Bub A, Ferrari P et al (1990) Urodilatin, a natriuretic factor from kidneys, can modify renal and cardiovascular function in men. Am J Physiol 259:F832–F838
Mitrovic V, Luss H, Nitsche K, Forssmann K, Maronde E, Fricke K et al (2005) Effects of the renal natriuretic peptide urodilatin (ularitide) in patients with decompensated chronic heart failure: a double-blind, placebo-controlled, ascending-dose trial. Am Heart J 150:1239
Cleland JGF, Coletta AP, Lammiman M, Witte KK, Loh H, Nasir M et al (2005) Clinical trials update from the European Society of Cardiology meeting 2005: CARE-HF extension study, ESSENTIAL, CIBIS-III, S-ICD, ISSUE-2, STRIDE-2, SOFA, IMAGINE, PREAMI, SIRIUS-II and ACTIVE. Eur J Heart Fail 7:1070–1075
Chen HH, Schirger JA, Alessandro C, Martin FL, Burnett JC (2006) Intra-renal infusion of BNP in experimental heart failure: A novel strategy to maximize the renal enhancing actions of BNP while minimizing arterial hypotension [abst]. J Card Fail 12:S32
Mathur VS, Goodson B, Patel S, Valencia A, Elkins J (2004) Evidence for substantial renal first pass effects of human b-type natriuretic peptide (nesiritide) following intra-renal infusion [abst]. J Card Fail 10:S68
Heywood JT, Ho A, Mathur V (2006) Favorable renal hemodynamic effects of intra-renal nesiritide infusion in heart transplant patients [abst]. J Card Fail 12:S3
Riter HG, Redfield MM, Burnett JC, Chen HH (2006) Nonhypotensive low-dose nesiritide has differential renal effects compared with standard-dose nesiritide in patients with acute decompensated heart failure and renal dysfunction [letter]. J Am Coll Cardiol 47:2334–2335
Luber JM Jr, The NAPA Investigators (2006) Perioperative nesiritide use is associated with decreased 180-day mortality in heart failure patients undergoing cardiothoracic surgery [abst]. J Card Fail 12:S73–S74
Oz MC. on behalf of the NAPA Investigators. Effect of perioperative nesiritide administration on postoperative renal function and clinical outcomes in patients undergoing cardiothoracic surgery: Results of the NAPA trial [abst]. Available at http://cicescardioorg/AbstractDetailsaspx?id = 28005 Accessed September 2, 2006
Zierer A, Safarzadeh E, Ewald GA, Pasque MK, Moon MR, Lawton JS et al (2006) Potential renal protective benefits of intra-operative BNP infusion during cardiac transplantation [abst]. Transplantation 82:564
Burnett JC Jr, Opgenorth TJ, Granger JP (1986) The renal action of atrial natriuretic peptide during control of glomerular filtration. Kidney Int 30:16–19
Brunner-La Rocca HP, Kaye DM, Woods RL, Hastings J, Esler MD (2001) Effects of intravenous brain natriuretic peptide on regional sympathetic activity in patients with chronic heart failure as compared with healthy control subjects. J Am Coll Cardiol 37:1221–1227
Bestle MH, Bie P (1993) Renal effects of urodilatin and atrial natriuretic peptide in volume expanded conscious dogs. Acta Physiol Scand 149:77–83
Chen HH, Grantham JA, Schirger JA, Jougasaki M, Redfield MM, Burnett JC Jr (2000) Subcutaneous administration of brain natriuretic peptide in experimental heart failure. J Am Coll Cardiol 36:1706–1712
Chen HH, Lainchbury JG, Harty GJ, Burnett JC Jr (2002) Maximizing the natriuretic peptide system in experimental heart failure: subcutaneous brain natriuretic peptide and acute vasopeptidase inhibition. Circulation 105:999–1003
Chen HH, Schirger JA, Cataliotti A, Burnett JC Jr (2006) Intact acute cardiorenal and humoral responsiveness following chronic subcutaneous administration of the cardiac peptide BNP in experimental heart failure. Eur J Heart Fail published online Feb 2, 2006
Chen HH, Huntley BK, Schirger JA, Cataliotti A, Burnett JC Jr (2006) Maximizing the renal cyclic 3′-5′-guanosine monophosphate system with type V phosphodiesterase inhibition and exogenous natriuretic peptide: A novel strategy to improve renal function in experimental overt heart failure. J Am Soc Nephrol 17:2742–2747
Chen HH, Redfield MM, Nordstrom LJ, Horton DP, Burnett JC Jr (2004) Subcutaneous administration of the cardiac hormone BNP in symptomatic human heart failure. J Card Fail 10:115–119
Madhani M, Okorie M, Hobbs AJ, Macallister RJ (2006) Reciprocal regulation of human soluble and particulate guanylate cyclases in vivo. Br J Pharmacol published online October 3, 2006
Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP (2006) NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov 5:755–768
Banga AK (2006) Oral delivery of peptide and protein drugs. Therapeutic peptides and proteins: formulation, processing, and delivery systems, 2nd edn. CRC Press, Boca Raton, FL, pp 229–258
Cataliotti A, Schirger JA, Martin FL, Chen HH, McKie PM, Boerrigter G et al (2005) Oral human brain natriuretic peptide activates cyclic guanosine 3′,5′-monophosphate and decreases mean arterial pressure. Circulation 112:836–840
Cataliotti A, Heublein DM, James KD, Burnett JC (2006) Biological actions of a novel oral human BNP in an experimental model of acute hypertension [abst]. J Card Fail 12:S30
Wang W, Ou Y, Shi Y (2004) AlbuBNP, a recombinant B-type natriuretic peptide and human serum albumin fusion hormone, as a long-term therapy of congestive heart failure. Pharm Res 21:2105–2111
Aaranson KD, Sackner-Bernstein JD (2006) Risk of death associated with nesiritide in patients with acutely decompensated heart failure. JAMA 296:1465–1466
Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K (2005) Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA 293:1900–1905
Sackner-Bernstein JD, Skopicki HA, Aaronson KD (2005) Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation 111:1487–1491
Braunwald E, Burnett JC Jr, Colucci WS, et al. Natrecor Advisory Panel Report. In: Panel of cardiology experts provides recommendations to Scios regarding Natrecor. Healthcare Professional Letter. Fremont, CA: Scios June 13, 2005. Available at www.natrecor.com/pdf/Braunwald_Panel_Release_&_Report.pdf Accessed September 12, (2006)
Panel of cardiology experts provides recommendations to Scios regarding Natrecor. Healthcare Professional Letter. Fremont, CA: Scios June 13, 2005. Available at www.natrecor.com/pdf/Braunwald_Panel_Release_&_Report.pdf Accessed September 12, (2006)
Biollaz J, Nussberger J, Porchet M, Brunner-Ferber F, Otterbein ES, Gomez H et al (1986) Four-hour infusions of synthetic atrial natriuretic peptide in normal volunteers. Hypertension 8:II96–II105
Kentsch M, Drummer C, Gerzer R, Muller-Esch G (1995) Severe hypotension and bradycardia after continuous intravenous infusion of urodilatin (ANP 95-126) in a patient with congestive heart failure. Eur J Clin Invest 25:281–283
Burnett J, Boerrigter G (2005) cGMP enhancing strategies for acute and chronic heart failure [abst]. BMC Pharmacology 5:S22
Grantham JA, Borgeson DD, Burnett JC Jr (1997) BNP: pathophysiological and potential therapeutic roles in acute congestive heart failure. Am J Physiol 272:R1077–R1083
Best PJ, Burnett JC, Wilson SH, Holmes DR Jr, Lerman A (2002) Dendroaspis natriuretic peptide relaxes isolated human arteries and veins. Cardiovasc Res 55:375–384
Collins E, Bracamonte MP, Burnett JC Jr, Miller VM (2000) Mechanism of relaxations to dendroaspis natriuretic peptide in canine coronary arteries. J Cardiovasc Pharmacol 35:614–618
Singh G, Maguire JJ, Kuc RE, Skepper JN, Fidock M, Davenport AP (2006) Characterization of the snake venom ligand [125I]-DNP binding to natriuretic peptide receptor-A in human artery and potent DNP mediated vasodilatation. Br J Pharmacol published online October 16, 2006
Lainchbury JG, Lisy O, Burnett JC Jr, Meyer DM, Redfield MM (2002) Actions of a novel synthetic natriuretic peptide on hemodynamics and ventricular function in the dog. Am J Physiol Regul Integr Comp Physiol 282:R993–R998
Lisy O, Jougasaki M, Heublein DM, Schirger JA, Chen HH, Wennberg PW et al (1999) Renal actions of synthetic dendroaspis natriuretic peptide. Kidney Int 56:502–508
Lisy O, Lainchbury JG, Leskinen H, Burnett JC Jr (2001) Therapeutic actions of a new synthetic vasoactive and natriuretic peptide, dendroaspis natriuretic peptide, in experimental severe congestive heart failure. Hypertension 37:1089–1094
Chen HH, Lainchbury JG, Burnett JC Jr (2002) Natriuretic peptide receptors and neutral endopeptidase in mediating the renal actions of a new therapeutic synthetic natriuretic peptide dendroaspis natriuretic peptide. J Am Coll Cardiol 40:1186–1191
Lewis RJ, Garcia ML (2003) Therapeutic potential of venom peptides. Nat Rev Drug Discov 2:790–802
Fry BG (2005) From genome to “venome”: molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res 15:403–420
Fry BG, Vidal N, Norman JA, Vonk FJ, Scheib H, Ramjan SF et al (2006) Early evolution of the venom system in lizards and snakes. Nature 439:584–588
Bazaa A, Marrakchi N, El Ayeb M, Sanz L, Calvete JJ (2005) Snake venomics: comparative analysis of the venom proteomes of the Tunisian snakes Cerastes cerastes, Cerastes vipera and Macrovipera lebetina. Proteomics 5:4223–4235
Barbouche R, Marrakchi N, Mansuelle P, Krifi M, Fenouillet E, Rochat H et al (1996) Novel anti-platelet aggregation polypeptides from Vipera lebetina venom: Isolation and characterization. FEBS Lett 392:6–10
Amininasab M, Elmi MM, Endlich N, Endlich K, Parekh N, Naderi-Manesh H et al (2004) Functional and structural characterization of a novel member of the natriuretic family of peptides from the venom of Pseudocerastes persicus. FEBS Lett 557:104–108
Michel GH, Murayama N, Sada T, Nozaki M, Saguchi K, Ohi H et al (2000) Two N-terminally truncated forms of C-type natriuretic peptide from habu snake venom. Peptides 21:609–615
Fry BG, Wickramaratana JC, Lemme S, Beuve A, Garbers D, Hodgson WC et al (2005) Novel natriuretic peptides from the venom of the inland taipan (Oxyuranus microlepidotus): isolation, chemical and biological characterisation. Biochem Biophys Res Commun 327:1011–1015
Ho PL, Soares MB, Maack T, Gimenez I, Puorto G, Furtado MF et al (1997) Cloning of an unusual natriuretic peptide from the South American coral snake Micrurus corallinus. Eur J Biochem 250:144–149
St Pierre L, Woods R, Earl S, Masci PP, Lavin MF (2005) Identification and analysis of venom gland-specific genes from the coastal taipan (Oxyuranus scutellatus) and related species. Cell Mol Life Sci 62:2679–2693
St Pierre L, Flight S, Masci PP, Hanchard KJ, Lewis RJ, Alewood PF et al (2006) Cloning and characterisation of natriuretic peptides from the venom glands of Australian elapids. Biochimie published online August 4, 2006
Murayama N, Hayashi MA, Ohi H, Ferreira LA, Hermann VV, Saito H et al (1997) Cloning and sequence analysis of a Bothrops jararaca cDNA encoding a precursor of seven bradykinin-potentiating peptides and a C-type natriuretic peptide. Proc Natl Acad Sci USA 94:1189–1193
Soares MR, Oliveira-Carvalho AL, Wermelinger LS, Zingali RB, Ho PL, Junqueira-de-Azevedo Ide L et al (2005) Identification of novel bradykinin-potentiating peptides and C-type natriuretic peptide from Lachesis muta venom. Toxicon 46:31–38
Ianzer D, Konno K, Marques-Porto R, Vieira Portaro FC, Stocklin R, Martins de Camargo AC et al (2004) Identification of five new bradykinin potentiating peptides (BPPs) from Bothrops jararaca crude venom by using electrospray ionization tandem mass spectrometry after a two-step liquid chromatography. Peptides 25:1085–1092
Hayashi MA, Camargo AC (2005) The Bradykinin-potentiating peptides from venom gland and brain of Bothrops jararaca contain highly site specific inhibitors of the somatic angiotensin-converting enzyme. Toxicon 45:1163–1170
Wei CM, Kim CH, Miller VM, Burnett JC Jr (1993) Vasonatrin peptide: a unique synthetic natriuretic and vasorelaxing peptide. J Clin Invest 92:2048–2052
Lisy O, Burnett JC Jr (2003) The design, synthesis and cardiorenal actions of a new chimeric natriuretic peptide CD-NP [abst]. J Am Coll Cardiol 41:312A
Lisy O, Burnett JC (2003) The new designer peptide CD-NP unloads the heart, suppresses renin and is natriuretic in vivo [abst]. J Card Fail 9:S32
Cataliotti A, Boerrigter G, Costello-Boerrigter LC, Schirger JA, Tsuruda T, Heublein DM et al (2004) Brain natriuretic peptide enhances renal actions of furosemide and suppresses furosemide-induced aldosterone activation in experimental heart failure. Circulation 109:1680–1685
Boerrigter G, Costello-Boerrigter LC, Lapp H, Stasch J-P, Burnett JC Jr (2005) Co-activation of soluble and particulate guanylate cyclase by BAY 58–2667 and BNP enhances cardiorenal function in experimental heart failure [abst]. J Card Fail 11(Suppl 1):S89
Costello-Boerrigter LC, Boerrigter G, Harty GJ, Burnett JC Jr (2006) Co-targeting of the V2 receptor with tolvaptan and the natriuretic peptide A-receptor with B-type natriuretic peptide enhances water and sodium excretion without adversely affecting renal function: A novel physiologic approach to sodium and water retention in experimental heart failure [abst]. J Card Fail 12:S84–S85
Hobbs AJ (2002) Soluble guanylate cyclase: An old therapeutic target re-visited. Br J Pharmacol 136:637–640
Friebe A, Koesling D (2003) Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res 93:96–105
Rothkegel C, Schmidt PM, Stoll F, Schroder H, Schmidt HH, Stasch JP (2006) Identification of residues crucially involved in soluble guanylate cyclase activation. FEBS Lett 580:4205–4213
Stasch JP, Schmidt P, Alonso-Alija C, Apeler H, Dembowsky K, Haerter M et al (2002) NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol 136:773–783
Ko FN, Wu CC, Kuo SC, Lee FY, Teng CM (1994) YC-1, a novel activator of platelet guanylate cyclase. Blood 84:4226–4233
Wu CC, Ko FN, Kuo SC, Lee FY, Teng CM (1995) YC-1 inhibited human platelet aggregation through NO-independent activation of soluble guanylate cyclase. Br J Pharmacol 116:1973–1978
Stasch JP, Becker EM, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A et al (2001) NO-independent regulatory site on soluble guanylate cyclase. Nature 410:212–215
Boerrigter G, Costello-Boerrigter LC, Cataliotti A, Tsuruda T, Harty GJ, Lapp H et al (2003) Cardiorenal and humoral properties of a novel direct soluble guanylate cyclase stimulator BAY 41-2272 in experimental congestive heart failure. Circulation 107:686–689
Stasch J-P, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, AK HS, Meurer S et al (2006) Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest 116:2552–2561
Scios announces the ETNA clinical trial with nesiritide: ETNA trial to be largest study of acute heart failure ever conducted. Press release September 21, 2005. Available at http://www.sciosinc.com/scios/pr_1127326387 Accessed September 29, 2006
Stiles S. Nesiritide shows hint of survival benefit in chronic-HF patients undergoing CABG. theheart.org. [HeartWire>Heart failure] September 12, 2006. Available at http://www.theheart.org/article/741265.do Accessed Sep 12, 2006
Baxter GF (2004) Natriuretic peptides and myocardial ischaemia. Basic Res Cardiol 99:90–93
Nieminen MS, Brutsaert D, Dickstein K, Drexler H, Follath F, Harjola V-P, et al (2006) EuroHeart Failure Survey II (EHFS II): a survey on hospitalized acute heart failure patients: description of population. Eur Heart J published online September 25, 2006:ehl193
Sezai A, Shiono M, Orime Y, Hata H, Hata M, Negishi N et al (2000) Low-dose continuous infusion of human atrial natriuretic peptide during and after cardiac surgery. Ann Thorac Surg 69:732–738
Wajima Z, Shiga T, Imanaga K, Inoue T, Ogawa R (2006) Effect of prophylactic bronchodilator treatment with i.v. carperitide on airway resistance and lung compliance after tracheal intubation. Br J Anaesth 96:660–664
Acknowledgments
Supported by grants from the National Institutes of Health (HL36634; PO1 HL76611 and HL80732) and the Mayo Foundation. Dr. Lee is supported by the Clinical Research Initiative Fellowship from the Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, C.Y.W., Burnett, J.C. Natriuretic peptides and therapeutic applications. Heart Fail Rev 12, 131–142 (2007). https://doi.org/10.1007/s10741-007-9016-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-007-9016-3