
Abstract
The inflammatory response plays an important role in carbon tetrachloride (CCl4)-induced acute liver injury and methane has been shown to exert beneficial effects on inflammation-associated diseases. Thus, we investigated the potential protective effects of methane-rich saline (MS) on CCl4-induced acute liver injury and explored the underlying mechanism. A CCl4-induced acute liver injury model was established by injection of CCl4 (0.6 ml/kg, ip) in mice followed by treatment with MS (16 ml/kg, ip), 24 h later. All groups of mice were sacrificed and blood and liver tissues were collected. Serum aminotransferase, necrotic areas, and inflammatory cell infiltration in liver slices were enhanced after CCl4 treatment but decreased with MS treatment. IL-6, TNF-α, IL-1β, IFN-γ, ICAM-1, CXCL1, MPO, NF-κB p65, ERK, JNK, and MAPK P38, expression in serum or liver homogenate were greater after CCl4 treatment but comparatively less after MS treatment. Only IL-10 increased after MS treatment. Anti-IL10 blockade (1.5 mg/kg) restored MS-mediated attenuated phosphorylation of NF-ĸbB/MAPK and the protective effect of MS was abolished for all indices examined. The PI3K inhibitor, wortmannin had the same effects on MS as anti-IL-10 antibody. MS also induced phosphorylation of GSK-3β and AKT in CCl4-treated mice. After pre-treatment with wortmannin (0.7 mg/kg), phosphorylation of GSK-3β and AKT proteins were reduced compared to its solvent control group-DMSO-treated animals. Thus, the data provide evidence that MS may activate the PI3K–AKT–GSK-3β pathway to induce IL-10 expression and produce anti-inflammatory effects via the NF-κB and MAPK pathways. The findings provide a new pharmacological strategy for management of inflammatory response after acute liver injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carbon tetrachloride (CCl4) is a hepatotoxicant that induces acute liver injury via generation of oxidative stress and recruitment of inflammatory cells (Mizuoka et al. 1999). CCl4-induced hepatotoxicity is characterized by sinusoidal congestion, neutrophil invasion, and ballooning degeneration and features abnormal serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) enzyme activity. Several pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) contribute to hepatic fibrosis (Abdel-Moneim et al. 2015; Peng et al. 2009), indicating that inflammation is central to CCl4-induced liver injury. CCl4-induced liver injury differs from other drug-induced liver injury such as Con A-induced hepatotoxicity which resembles autoimmune liver disease.
Interleukin-10 (IL-10) is an anti-inflammatory pleiotropic cytokine which suppresses effector cells and mediates release of multiple cytokines (Si et al. 2010) to decrease T lymphocyte activity (Martin et al. 2010; Douglas et al. 2010) and strengthen immune tolerance. Previous work indicated that IL-10 can suppress ConA-induced liver injury (Erhardt et al. 2007; Shao et al. 2013) and ischemia–reperfusion injury (Ren et al. 2011). Dysregulated IL-10 production also contributes to inflammatory diseases (Ouyang et al. 2011; Yao et al. 2013).
The mechanism that controls IL-10 production in response to specific stimuli has been shown to include MAPK (Hovsepian et al. 2013) and several transcription factors. Nuclear factor ĸB (NF-ĸB) and MAPK regulate expression of many genes critical to the regulation of apoptosis, viral replication, tumorigenesis, inflammation, and various autoimmune diseases. Activation of NF-ĸB and MAPK may be a component of the stress response because they were activated by diverse stimuli including growth factors, cytokines, lymphokines, UV, pharmacological agents, and stress. Our work suggests that MS reduced phosphorylation of NF-ĸB, JNK, ERK and p38 in LPS-stimulated macrophages in an IL-10 dependent manner via enhanced activation of PI3K/AKT signal which involved activated GSK-3β (Zhang et al. 2016). Inhibition of GSK-3β was reported to reduce liver ischemia reperfusion injury via an IL-10-mediated immunoregulatory mechanism (Ren et al. 2011). Thus, we studied the relationship between PI3K–AKT–GSK-3β pathways and mechanisms of IL-10 production.
Methane is the most common organic atmospheric gas and fuel source (Montano-Loza et al. 2017). Previous studies focused on biological characteristics of methane in the gastrointestinal tract (Ghoshal et al. 2016), specifically finding that exogenously applied methane has protective effects on the intestine (Varga et al. 2012; Boros et al. 2012), heart (Chen et al. 2015), abdominal skin flaps (Song et al. 2015), retina (Liu et al. 2016; Wu et al. 2015), and nervous system (Fan et al. 2016; Wang et al. 2016). Hepatoprotection is also reported to be conferred by methane (Strifler et al. 2016; Ye et al. 2015; He et al. 2016). Methane has been shown to inhibit the inflammatory response via modulating various pathways. Studies of methane include physiological saline for gas dissolution to reduce flammability and explosiveness of methane gas (Zhang et al. 2016). Methane gas is relatively stable for 1 month (Roccarina et al. 2010; Chen et al. 2015) and as such may have therapeutic use. Therefore, we studied the anti-inflammatory and protective effects of methane in a CCl4-induced acute liver injury model by measuring cytokine IL-10 and associated intracellular signaling pathways.
Materials and methods
Animals and reagents
C57BL/6 mice (6–8 weeks-of-age; 18–22 g; male), were purchased from the Animal Experimentation Center of the Second Military Medical University. All animals were housed under specific pathogen-free conditions and were provided with Rodent Lab Chow and water ad libitum. CCl4 was purchased from Sinopharm Chemical Reagent Co., Ltd (China) and was dissolved in olive oil. All other chemicals and reagents used were standard analytical grade.
Methane-rich saline production
Methane was stored in a gas canister and was dissolved in normal saline under high pressure (0.4 MPa) for 3 h to a saturated level. The saturated MS was stored under atmospheric pressure at 4 °C and was freshly prepared 1 day prior to the animal experiments to ensure stability. Gas chromatography (gas chromatography-9860, Qiyang Co., Shanghai, China) was used to measure methane in the saline solution according to published methods (Ren et al. 2011).
CCl4-induced acute liver injury in mice
Mice were pretreated with DMSO, anti-IL10 antibody (1.5 mg/kg) or wortmannin (0.7 mg/kg) for 24 h. Acute liver injury was induced by injecting CCl4 (0.6 ml/kg, ip, 12 μl:400 µl olive oil). MS (16 ml/kg) was administered 1 h after CCl4 injection, and sham mice were treated with olive oil (16 ml/kg). Serum and liver specimens were collected at the indicated time points.
Measurement of liver enzymes and cytokine production
Mice were anesthetized 24 h after CCl4 treatment and blood was collected via heart puncture. Plasma was separated following centrifugation at 300×g for 10 min. Serum ALT and AST were measured as described by Magaye et al. (2016) with an automatic dry biochemical analyzer (Hitachi Auto Analyzer 7170, Japan). Serum IL-6, TNF-α, IL-1β, IFN-γ and IL-10 and myeloperoxidase (MPO), chemokine ligand 1 (CXCL1), Intercellular adhesion molecule-1 (ICAM-1) in liver homogenates were measured using ELISA (eBioscience, San Diego, CA) according to the manufacturer’s instructions.
Histopathological assessment
Liver tissues were harvested in 24 h after CCl4 administration. Liver samples were preserved in 4% paraformaldehyde for a minimum period of 72 h. Specimens were embedded in paraffin and cut into 4–5 µm sections for hematoxylin and eosin (H&E) staining. Inflammation and tissue damage were observed using light microscopy.
Real-time PCR
Total RNA was extracted from liver tissue using Trizol reagent (Takara, Japan). Concentration and purity of total RNA was measured using the absorbance ratios at 260/280 nm. Complementary DNA (cDNA) was reverse-transcribed using a Prime Script RT Reagent Kit (Takara). Quantification of IL-6, TNF-α, IL-1β, IFN-γ, CXCL1, ICAM-1 and IL-10 mRNA was conducted using Step One Plus Real Time PCR System (Applied Biosystems, CA). Murine primers were synthesized by Invitrogen and sequences were as follows:
-
IL-6:F5′-TACCACTCCCAACAGACCTG-3′(forward),5′-GGTACTCCAGAAACCAGAGG-3′(reverse);TNF-α:5′-CACCATGAGCACAGAAAGCA-3′(forward), 5′-TAGACAGAAGAGCGTGGTGG-3′(reverse);IL-1β:5′-ACTCATTGTGGCTGTGGAGA-3′(forward),5′-TTGTTCATCTCGGAGCCTGT-3′(reverse);IFN-γ:5′-CCTCAAACTTGGCAATACTCA-3′(forward);5′-CTCAAGTGGCATAGATGTGGA-3′(reverse);CXCL1:5′-GCTTGAAGGTGTTGCCCTCAG-3′(forward),5′-AGAAGCCAGCGTTCACCAGAC-3′(reverse);ICAM-1:5′-TTCACACTGAATGCCAGCCC-3′(forward);5′-GTCTGCTGAGACCCCTCTTG-3′(reverse);IL-10:5′-TGCCACTCAGAAGACTGYGG-3′(forward),5′-GTCCTCAGTGTAGCCCAGGA-3′(reverse);GAPDH:5′-ATGGTGAAGGTCGGTGTGAA-3′(forward),5′-TGGAAGATGGTGATGGGCTT-3′(reverse).
Western blotting
Liver tissues from experimental animals were homogenized in protein lysis buffer (Thermo Fisher Scientific, location missing) with protease inhibitor (Gibco, location missing). After centrifugation (13,000×g, 4 °C, 10 min), protein was measured using a BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of protein were loaded in each well and separated using 10% SDS-PAGE (Life Technologies, Carlsbad, CA). Gels were subsequently transferred to nitrocellulose membranes (Life Technologies) and membranes were blocked for 1 h in 5% non-fat dried milk and then incubated overnight at 4 °C with primary antibodies (Cell Signaling Technology). Membranes were washed with TBST and incubated with secondary antibody for 2 h at room temperature. Finally, protein bands were visualized using an ECL kit (Thermo Fisher Scientific, Waltham, MA). Band intensity was quantified using Image J software and protein expression was normalized according to expression of GAPDH protein.
Statistical analysis
Data are presented as means ± standard deviation (SD). Significant differences were confirmed with one-way ANOVA, followed by Turkey’s test and two-way ANOVA. Prism 5 software package (GraphPad) was used to calculate p values and p < 0.05 was considered statistically significant.
Results
MS protects mice from CCl4-induced liver injury and inhibits CCl4-induced liver inflammation
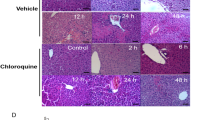
Serum ALT and AST activity significantly increased after CCl4 treatment and MS prevented this elevation (Fig. 1C, D). Histopathological studies supporting the biochemical data (Fig. 1A). Liver sections obtained from sham and treatment (sham + MS) groups had normal liver architecture (a, b, g, h) and no necrosis (Fig. 1B). CCl4 caused large areas of extensive pericentral necrosis (Fig. 1B) with hepatic sinus congestion, neutrophil infiltration, ballooning degeneration, and loss of hepatic architecture (c, i). After MS administration to mice, the development of histopathological alterations induced by CCl4 were inhibited (d, j), and necrosis was decreased (Fig. 1B). Data suggested that MS protected mice against CCl4-induced liver injury.

MS protects mice from CCl4-induced liver injury. Liver tissues were stained with H&E after collection from mice sacrificed 24 h after treatment with sham (a, g), sham + MS (b, h), CCl4 (c, i), CCl4 + MS (d, j), CCl4 + MS + anti-IL10 (e, k), CCl4 + MS + wortmannin (f, l) (Scale bar 100 µm, 50 µm), Arrow indicates preserved hepatic sinus congestion and ballooning degeneration; squares indicate areas of neutrophil infiltration and loss of hepatic architecture (A), necrotic areas were analyzed (B). Serum ALT and AST for each group (C, D). Data are as means ± SD (n = 6). *p < 0.05, **p < 0.01 vs. sham group, # p < 0.05, ## p < 0.01 vs. CCl4 group, Δ p < 0.05, ΔΔ p < 0.01 vs. CCl4 + MS group
Serum cytokines were measured after treatment with CCl4 and IL-6, TNF-α, IL-1β and IFN-γ were elevated and MS treatment reduced these elevations (Fig. 2a–d). Relative IL-6, TNF-α, IL-1β and IFN-γ mRNA was significantly increased after CCl4 treatment but decreased after MS treatment (Fig. 2e–h). Thus, MS protects liver function by inhibiting expression of inflammatory cytokines. We also measured neutrophils such as CXCL1, MPO and ICAM-1. Serum CXCL1, ICAM-1 and MPO activity and mRNA were increased in CCl4-treated mice and decreased after treatment with MS (Fig. 2i–m). IL-10 expression and mRNA was not significantly changed after CCl4 treatment but it was increased with CCl4 and MS administration (Fig. 2n, o). Thus, anti-inflammatory effects of MS may be tied to IL-10.

MS inhibits inflammation in CCl4-induced acute liver injury in mice. Serum IL-6, TNF-α, IL-1β, IFN-γ, IL-10 (a–d, n) and liver homogenate ICAM-1 (i), CXCL1 (k) were measured using ELISA. IL-6, TNF-α, IL-1β, IFN-γ (e–h), ICAM-1 (j), CXCL1 (l) and IL-10 (o) mRNA were measured using RT-PCR. MPO activity quantification in liver homogenates (m). Data are means ± SD (n = 6), *p < 0.05, **p < 0.01 vs. sham group, # p < 0.05, ## p < 0.01 vs. CCl4 group, Δ p < 0.05, ΔΔ p < 0.01 vs. CCl4 + MS group
Anti-IL-10 antibody and wortmannin reversed protective effects of MS in CCl4-induced acute liver injury
To study the contribution of IL-10 to the hepatoprotective effect of MS, we used an anti-IL-10 antibody and measured liver function. After CCl4 and MS treatment with the antibody, serum ALT and AST were restored to normal (Fig. 1C, D). Similarly, histopathological studies indicated that the inhibitory effect of MS was partially reduced in mice in the presence of the anti-IL-10 antibody (Fig. 1A, B). Serum inflammatory cytokines (Fig. 2a–h) and neutrophil chemokines (Fig. 2i–m) and their mRNA were increased with concomitant administration of the anti-IL-10 antibody and MS. Thus, MS produced anti-inflammatory effects and restored liver function in CCl4-treated mice via increased production of IL-10. GSK-3β is associated with IL-10 secretion via modulation of the PI3K–AKT pathway, so we inhibited GSK-3β with wortmannin and data show that the protective effect of MS was blocked.
MS reduces activation of NF-κB and MAPK in CCl4-induced liver injury
The NF-κB and MAPK pathways are responsible for inflammatory cytokine production in mammals so whether MS influenced expression of NF-κB p65, ERK, JNK and MAPK P38 proteins was investigated and data show that NF-κB, ERK, JNK and p38 signaling pathways were activated by CCl4. MS treatment inhibited phosphorylation of NF-κB P65, ERK, JNK and p38 proteins (Fig. 3a) and these results were statistically significant (Fig. 3c–f).

Anti-IL-10 antibody and wortmannin reversed NF-κB and MAPK protein phosphorylation in MS-treated mice according to Western blot of liver homogenates (a, b). Band intensities were quantified as ratios of phosphorylated signaling molecules to total molecules (c–n). *p < 0.05, **p < 0.01
Anti-IL-10 and wortmannin reverses reduced NF-κB protein and MAPK phosphorylation in MS-treated mice
Studies suggest that GSK-3 acts on IL-10 and influences the NF-κB pathway. To explore anti-inflammatory mechanism of MS, mice were pretreated with an anti-IL-10 antibody and wortmannin as previously described. NF-κB and MAPK protein expression was measured before and after inhibition of PI3K enzyme and IL-10 (Fig. 3b). Pretreatment anti-IL-10 antibody and wortmannin increased the ratio of phosphorylated to total the NF-κB and MAPK proteins (Fig. 3g–n) in MS-treated mice, suggesting that these proteins regulate anti-inflammatory processes in CCl4-treated mice. So, anti-IL-10 antibody and wortmannin abolished the inhibitory effect of MS via increased phosphorylation of MAPK proteins and NF-κB.
IL-10 expression was mediated by the PI3K–AKT–GSK-3β pathway in MS-treated mice
MS diminished expression of pro-inflammatory factors and protected livers against injury induced by CCl4. MS augmented expression of IL-10 and inhibited the phosphorylation of NF-κB, ERK, JNK, and p38 (Figs. 1, 2, 3). Anti-IL-10 antibody and wortmannin treatment reversed hepatoprotection offered by MS. Thus, an association between IL-10 and PI3K was investigated (Fig. 4) and treatment with MS increased phosphorylation of GSK-3β and AKT after CCl4-induced liver injury and wortmannin treatment inhibited activation of GSK-3β and AKT (Fig. 4). Wortmannin treatment decreased IL-10 (Fig. 2n, o), suggesting that GSK-3β was essential for IL-10 expression. This, data suggest that MS activates the PI3K–AKT pathway and promotes IL-10 expression via activation of GSK-3β which then produces an anti-inflammatory effect via the NF-κB and MAPK pathways.

GSK-3β-mediated IL-10 expression in MS-treated mice is promoted via the activation of PI3K–AKT. Mice were pre-treated with DMSO or wortmannin (0.7 mg/kg) for 24 h and Western blot was used to quantify protein (a, c). Band intensities were quantified as ratios of phosphorylated signaling proteins to total proteins (b, d). **p < 0.01 vs. sham group, ΔΔ p < 0.01 vs. CCl4 + MS group
Discussion
Methane is one of the simplest organic compounds (Zhang et al. 2016) and it is increasingly being studied for medical applications. We reported that MS protected against LPS-induced inflammation and ConA-induced liver injury in several animal models (Zhang et al. 2016; He et al. 2016) and here we report that MS was protective against CCl4-induced acute liver injury. CCl4 caused extensive necrotic areas and inflammatory infiltration in mouse liver sections. Serum ALT and AST were used to establish the severity of liver injury (Maes et al. 2016; Szabo and Petrasek 2015). Treatment with MS significantly decreased necrotic areas and inflammatory infiltration and reduced serum ALT and AST. Thus, MS reduced liver damage induced by CCl4.
Tissue inflammation contributes to liver pathology and pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, IFN-γ are associated with the pathogenesis of drug-induced liver injury (Mackenzie et al. 2013; Juhaszova et al. 2004). We hypothesized that MS exerted effects by reducing inflammation so we measured IL-6, TNF-α, IL-1β and IFN-γ mRNA and protein and found that they were increased in CCl4-treated mice. Treatment with MS after CCl4 administration reduced elevations IL-6, TNF-α, IL-1β and IFN-γ. CXCL1, MPO and ICAM-1 expression are key to the migration of neutrophils (Özdemir-Kumral et al. 2016; Zhao et al. 2016) and MS treatment decreased chemokines protein and mRNA, confirming anti-inflammatory effects of MS in our mouse model.
IL-10 blocks the inflammatory response (Bekker et al. 2016; Yao et al. 2013a, b) and may protect against hepatic injury (Peng et al. 2009; Rood et al. 2016). MS treatment increased IL-10 protein and mRNA expression and inhibition of IL-10 expression with an anti-IL-10 antibody reduced elevated ALT and AST, pro-inflammatory cytokines and neutrophilic chemotactic factor. Thus, IL-10 may protect the liver from injury.
IL-10 is reported to modulate the inflammatory response by inhibiting NF-κB, ERK/MAPK pathways (Peng et al. 2009; Mittal and Roche 2015; Gabrysova et al. 2014), and transcription factors such as CREB, NF-κB p50 homodimers, and C/EBPb (Ananieva et al. 2008; Cao et al. 2006; Nandan et al. 2012; MacKenzie et al. 2013; Sanin et al. 2015). However, we did not observe upregulation of activation of p38MAPK in MS-treated mice. To investigate the down-stream mechanism of IL-10, we neutralized IL-10 expression with an anti-IL-10 antibody and measured phosphorylation of proteins activated by NF-κB transcription factor and MAPK. Data show that neutralization of IL-10 partially reversed MS-induced decreased NF-κB p65, ERK, JNK and P38 protein phosphorylation. Thus, IL-10 may inhibit pro-inflammatory cytokine expression via the NF-κB and MAPK pathway in MS-treated mice.
The PI3K–AKT–GSK-3β pathway is thought to be a significant producer of IL-10 (Beurel et al. 2010, 2015) and the PI3K inhibitor wortmannin inhibited protective and anti-inflammatory effects of MS after liver injury induced by CCl4. MS treatment increased IL-10 and wortmannin decreased them. MS increased and prolonged expression of p-GSK-3β and p-AKT, and PI3K inhibition reduced these effects indicating involvement of the PI3K–AKT–GSK-3β pathway in MS-induced IL-10 production.
Akt is activated by phosphorylation of various enzymes, kinases, transcription factors and other downstream factors and this downregulates IL-10. The mammalian target of rapamycin (mTOR), activation of GSK-3β, and phosphorylation of Foxo138-40, so mTOR and Foxo1 may also act upstream to mediate p-GSK-3β-induced IL-10 production in MS-treated mice.
IκB kinase (IKK) is activated by Akt and this leads to the degradation of NF-κβ inhibitor, Iκβ, and enhances expression of NF-κB and promotes cell survival. As NF-κB was not upregulated in MS-treated mice, how MS exerts effects on the PI3K–AKT pathway is unclear, but it may act on membrane channels of G-proteins, membrane receptor-mediated signaling, or acetylcholine-activated ion channel kinetics (Kai et al. 1998; Sokoll et al. 1989; Puig et al. 1988). Alternatively, MS may accumulate at interfaces of cell membranes to modulate the function of membrane-bound enzymes (Ghyczy et al. 2008). MS easily penetrates membranes and diffuses into organelles (Pimentel et al. 2006; Venardos et al. 2007), so it may penetrate and activate PI3K–AKT, a hypothesis that supports our observation MS peaked in the circulation of mice in 10 min.
The molecular mechanism underlying GSK-3β regulation of anti-inflammatory cytokine expression in our study is poorly characterized. Active GSK-3β may reduce NF-κΒ activation by enhancing interactions of CREB with the CBP, which leads to reduced CBP binding with NF-κΒ (53). Alternatively, GSK-3β may facilitate NF-κΒ activity by activation of NF-κΒ p65 (Viatour et al. 2004) and limitation of NF-κΒ activation in unidentified pathways (Schwabe et al. 2002). It is more likely that reduced activation of NF-κΒ and p38 MAPK contributes to GSK-3β-mediated IL-10 production in our study, because neutralization of IL-10 partially reversed decreased activation of NF-κΒ and MAPK. Because the intracellular signaling pathways controlling IL-10 production are complex, how p-GSK-3β regulates IL-10 expression in our study was not resolved.
In conclusion, MS protected against CCl4-induced acute liver injury as evidenced by liver function enzymes and reduced liver injury. MS treatment significantly inhibited inflammatory responses likely via activation of the PI3K–AKT–GSK-3β pathway and increased production of IL-10 and suppressed NF-κB and MAPK signaling in CCl4-treated mice. Furthermore, inhibition of IL-10 and GSK-3β reduced the protective effects of MS in CCl4-treated mice, suggesting a requirement of IL-10 for this signaling pathway and the contribution of IL-10 production to the protective effect of MS.
Our study has some limitations. CCl4-induced liver injury has been confirmed in neutrophils and Kupffer cells so in future studies, we will focus on IL-10 activity in different cell types. Also, different time points and a MS concentration gradient are needed to understand dose and time-responses of MS for hepatic protection against acute injury. We must clarify whether hepatoprotective effect is restricted to CCl4 hepatotoxicity as well. Despite the aforementioned limitations, MS offers a new therapeutic strategy for clinical application and our data suggest a rationale for developing new pharmacological strategies to treat acute liver injury.
References
Abdel-Moneim AM, Al-Kahtani MA, El-Kersh MA et al (2015) Free radical-scavenging, anti-inflammatory/anti-fibrotic and hepatoprotective actions of taurine and silymarin against CCl4 induced rat liver damage. PLoS ONE 10:e0144509
Ananieva O et al (2008) The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat Immunol 9:1028–1036
Bekker Z, Walubo A, Du Plessis JB et al (2016) Changes in IL-2 and IL-10 during Chronic Administration of Isoniazid, Nevirapine, and Paracetamol in Rats. Adv Pharmacol Sci 11:3094783
Beurel E, Michalek SM, Jope RS et al (2010) Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol 31:24–31
Beurel E, Grieco SF, Jope RS et al (2015) Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148:114–131
Boros M, Ghyczy M, Érces D et al (2012) The anti-inflammatory effects of methane. Crit Care Med 4:1269–1278
Cao S, Zhang X, Edwards JP, Mosser DM (2006) NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem 281:26041–26050
Chen O, Ye Z, Cao Z et al (2015) Methane attenuates myocardial ischemia injury in rats through anti-oxidative, anti-apoptoticand anti-inflammatory actions, Free Radic Biol Med 90:1–11
Douglas DB, Beiting DP, Loftus JP et al (2010) Combinatorial effects of interleukin 10 and interleukin 4 determine the progression of hepatic inflammation following murine enteric parasitic infection. Hepatology 51:2162–2171
Erhardt A, Biburger M, Papadopoulos T et al (2007) IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology 45:475–485
Fan DF, Hu HJ, Sun Q et al (2016) Neuroprotective effects of exogenous methane in a rat model of acute carbon monoxide poisoning. Brain Res 1633:62–72
Gabrysova L, Howes A, Saraiva M et al (2014) The regulation of IL-10 expression. Curr Top Microbiol Immunol 380:157–190
Ghoshal U, Shukla R, Srivastava D et al (2016) Irritable bowel syndrome, particularly the constipation-predominant form, involves an increase in methanobrevibacter smithii, which is associated with higher methane production. Gut Liver 6:932–938
Ghyczy M et al (2008) Hypoxia-induced generation of methane in mitochondria and eukaryotic cells: an alternative approach to methanogenesis. Cell Physiol Biochem 21:251–258
He R, Wang L, Zhu J et al (2016) Methane-rich saline protects against concanavalin A-induced autoimmune hepatitis in mice through anti-inflammatory and anti-oxidative pathways. Biochem Biophys Res Commun 1:22–28
Hovsepian E, Siffo S, Mirkin GA et al (2013) IL-10 inhibits the NF-ĸB and ERK/MAPK-mediated production of pro-inflammatory mediators by up-regulation of SOCS-3 in Trypanosoma cruzi-infected cardiomyocytes. PLoS ONE 8:e79445
Juhaszova M, Zorov DB, Kim SH et al (2004) Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113:1535–1549
Kai T, Jones KA, Warner DO et al (1998) Halothane attenuates calcium sensitization in airway smooth muscle by inhibiting G-proteins. Anesthesiology 89:1543–1552
Liu L, Sun Q, Wang R et al (2016) Methane attenuates retinal ischemia/reperfusioninjury via anti-oxidative and anti-apoptotic pathways. Brain Res 1646:327–333
Mackenzie KF, Clark K, Naqvi S, Mcguire VA et al (2013) PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J Immunol 190:565–577
MacKenzie KF et al (2013) PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J Immunol 190:565–577
Maes M, Vinken M, Jaeschke H et al (2016) Experimental models of hepatotoxicity related to acute liver failure. Toxicol Appl Pharmacol 290:86–97
Magaye R, Gu Y, Wang Y et al (2016) In vitro and in vivo evaluation of the toxicities induced by metallic nickel nano and fine particles. J Mol Histol 47:273–286
Martin M, Rehani K, Jope RS et al (2005) Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 6:777–784
Martin M, Mory C, Prescher A et al (2010) Protective effects of early CD4(+) T cell reduction in hepatic ischemia/reperfusion injury. J Gastrointest Surg 14:511–519
Mittal SK, Roche PA (2015) Suppression of antigen presentation by IL-10. Curr Opin Immunol 34:22–27
Mizuoka H, Shikata N, Yang J et al (1999) Biphasic effect of colchicine on acute liver injury induced by carbon tetrachloride or by dimethylnitrosamine in mice. J Hepatol 31:825–833
Montano-Loza AJ, Bhanji RA, Wasilenko S et al (2017) Systematic review: recurrent autoimmune liver diseases after liver transplantation. Aliment Pharmacol Ther 45:485–500
Nandan D et al (2012) Myeloid cell IL-10 production in response to leishmania involves inactivation of glycogen synthase kinase-3beta downstream of phosphatidylinositol-3 kinase. J Immunol 188:367–378
Ouyang W, Rutz S, Crellin NK et al (2011) Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol 29:71–109
Özdemir-Kumral ZN, Özbeyli D, Özdemir AF et al (2016) Protective effect of nicotine on sepsis-induced oxidative multiorgan damage: role of neutrophils. Nicotine Tob Res 2017(7):859–864
Peng XD, Dai LL, Huang CQ et al (2009) Relationship between anti-fibrotic effect of Panax notoginsengsaponins and serum cytokines in rat hepatic fibrosis. BiochemBiophys Res Commun 388:31–34
Pimentel M et al (2006) Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am J Physiol Gastrointest Liver Physiol 290:G1089–G1095
Puig MM, Warner W, Tang CK, Lovitz M, Turndorf H et al (1988) Synergistic interaction of morphine and halothane in the guinea pig ileum. Anesthesiology 68:559–562
Ren F, Duan Z, Cheng Q et al (2011) Inhibition of glycogen synthase kinase 3 beta ameliorates liver ischemia reperfusion injury by way of an interleukin-10-mediated immune regulatory mechanism. Hepatology 54:687–696
Roccarina D, Lauritano EC, Gabrielli M et al (2010) The role of methane in intestinal diseases. Am J Gastroenterol 105:1250–1256
Rood JE, Canna SW, Weaver LK et al (2016) IL-10 distinguishes a unique population of activated, effector-like CD8+ T cells in murine acute liverinflammation. J Leukoc Biol 2017(4):1037–1044
Sanin DE, Prendergast CT, Mountford AP et al (2015) IL-10 production in macrophages is regulated by a TLR-driven CREB-mediated mechanism that is linked to genes involved in cell metabolism. J Immunol 195:1218–1232
Schwabe RF, Brenner DA et al (2002) Role of glycogen synthase kinase-3 in TNF-alpha-induced NF-kappaB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol 283:G204–G211
Shao X, Qian Y, Xu C et al (2013) The protective effect of intrasplenic transplantation of Ad-IL-18BP/IL-4 gene-modified fetal hepatocytes on ConA-induced hepatitis in mice. PLoS ONE 8:e58836
Si ZZ, Li JQ, Qi HZ et al (2010) Recombinant adenovirus vector AdhIL-10 protects grafts from cold ischemia-reperfusion injury following orthotopic liver transplantation in rats. Hepatobiliary Pancreat Dis Int 9:144–148
Sokoll MD, Davies LR, Bhattacharyya B, Zwagerman DQ et al (1989) Halothane and isoflurane alter acetylcholine activated ion channel kinetics. Eur J Pharmacol 173:27–34
Song K, Zhang M, Hu J et al (2015) Methane-rich saline attenuates ischemia/reperfusion injury of abdominal skin flaps in rats via regulating apoptosis level. BMC Surg 15:92
Strifler G, Tuboly E, Szél E et al (2016) Inhaled methane limits the mitochondrial electron transport chain dysfunction during experimental liver ischemia-reperfusion injury. PLoS ONE 1:e0146363
Szabo G, Petrasek J (2015) Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol 7:387–400
Varga G, Erces D, Tuboly E et al (2012) Characterization of the anti-inflammatory properties of methane inhalation during ischaemia-reperfusion. Magy Seb 4:205–211
Venardos KM, Perkins A, Headrick J, Kaye DM et al (2007) Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: a review. Curr Med Chem 14:1539–1549
Viatour P et al (2004) GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell 16:35–45
Wang L, Yao Y, He R et al (2016) Methane ameliorates spinal cord ischemia-reperfusion injury in rats: antioxidant, anti-inflammatory and anti-apoptotic activity mediated by Nrf2 activation. Free Radic Biol Med 103:69–86
Wu J, Wang R, Ye Z et al (2015) Protective effects of methane-rich saline on diabetic retinopathy via anti-inflammation in a streptozotocin-induced diabetic rat model. Biochem Biophys Res Commun 2:155–161
Yao Y, Simard AR, Shi FD et al (2013a) IL-10-producing lymphocytes in inflammatory disease. Int Rev Immunol 32:324–336
Yao Y, Simard AR, Shi F (2013b) D.et al, IL-10-producing lymphocytes in inflammatory disease. Int Rev Immunol 32:324–336
Ye Z, Chen O, Zhang R et al (2015) Methane attenuates hepatic ischemia/reperfusion injury in rats through antiapoptotic, anti-inflammatory, and antioxidative actions. Shock 44:181–187
Zhang X, Li N, Shao H et al (2016) Methane limit LPS-induced NF-ĸB/MAPKs signal in macrophages and suppress immune response in mice by enhancing PI3K/AKT/GSK-3β-mediated IL-10 expression. Sci Rep 6:29359
Zhao X, Shi X, Zhang Z et al (2016) Combined treatment with MSC transplantation and neutrophil depletion ameliorates D-GalN/LPS -induced acute liver failure in rats. Clin Res Hepatol Gastroenterol 6:730–738
Acknowledgements
This work was supported by National Natural Science Foundation (81471845, 81671939) and Shanghai Scientific research project (15411963200).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
All authors have no potential financial or ethical conflicts of interest regarding this paper.
Additional information
Ying Yao, Liping Wang and Peipei Jin have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yao, Y., Wang, L., Jin, P. et al. Methane alleviates carbon tetrachloride induced liver injury in mice: anti-inflammatory action demonstrated by increased PI3K/Akt/GSK-3β-mediated IL-10 expression. J Mol Hist 48, 301–310 (2017). https://doi.org/10.1007/s10735-017-9728-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10735-017-9728-1