
Abstract
Germplasm collections of cultivated plants constitute the source for further genetic progress. These collections gained further interest with approaches for tracking allelic variants associated to phenotypic variations within core collections. In order to explore the structure of genetic variation in pepper (Capsicum spp.) and to select core-collections maximizing the genetic and the phenotypic diversity, a pepper collection including 1,352 non redundant accessions from 11 Capsicum species from 89 different countries was genotyped using 28 microsatellite (SSR) markers spanning the whole genome. Model-based analysis structured the collection into 6 clusters, with 3 clusters separating the main species complexes, including cultivated species and wild relatives, according to taxonomic classification (C. frutescens/C. chinense, C. baccatum, C. pubescens), and 3 additional clusters for C. annuum. The relationships between the cultivated C. annuum species and the wild relative (C. annnuum var. glabriusculum) was precised. The 3 C. annuum clusters were significantly distinct for plant and fruit descriptors corresponding to cultivar types, showing that the genetic structure of this cultivated species was strongly impacted by the long term human selection of landraces in primary as well as secondary diversification centres. We settled nested core-collections of 8, 16, 32, 64 and 128 C. annuum accessions capturing from 37 to 90 % of the genetic diversity for further sequencing efforts and establishment of high throughput genotyping assays. By compiling phenotypic and genotypic data, a larger core-collection of 332 accessions was established, capturing 97 % of the C. annuum genetic and phenotypic diversity for further genetic association studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In plant breeding, genetic diversity is the essential source of genetic progress. This motivated the collection, maintenance and characterization of genetic resources for most cultivated plant species by private, national and international initiatives since the early 20th century. These plant Germplasm libraries of various sizes include wild accessions, landraces, modern cultivars and related species of the crop of interest. Efforts in securing plant genetic resources for food and agriculture recently progressed worldwide, since it is regarded as an essential resource to help farmers to respond to climate change (FAO 2010). Safeguarding wild accessions is an increasing priority to prevent the loss of wild genotypes resulting from the reduction of natural wild habitats worldwide. Conservation of landraces and local cultivars is as well crucial, since modern and highly performing cultivars progressively replace the diversified and heterogeneous landraces worldwide, threatening the agricultural landscape with genetic erosion (Hammer et al. 2003). For most cultivated species, the loss of genetic variability started as soon as the domestication process, and may have been worsened with migration events from original to secondary diversification sites depending on the history of plant species exploitation (Tang et al. 2010). Contrarily, thousands of years of human selection in multiple environments and cultural contexts, provided new mutants and allele combinations of agricultural interest but which had poor probabilities to be retained under natural selection pressure. The thousands of landraces and local cultivars originating from farmer selection in each crop represent a wide source of diversity, particularly for alleles of agricultural interest and local adaptations (i.e. quality traits, tolerance or resistance to biotic and abiotic stresses). Their contribution to further genetic progress and to the restoration of biodiversity in agro systems is at least as promising as wild accessions, related species or exogenous gene sources.
Beyond the collection of these resources, their exploitation depends on our ability to characterize them. The exhaustive phenotyping and genotyping of large collections has rarely been performed. Within the plant breeding process, large screening tests focus on a particular trait in order to trap the alleles of interest. Then, segregating progenies are generated for genetic analysis of the trait and the new alleles are further introgressed into elite genitors in a cumulative genetic progress. More recently, approaches were developed to track the allelic variants associated to phenotypic variations directly within core collections of plant genotypes, i.e. subsamples of genotypes which represent the genetic diversity of the crop with a minimal redundancy (Marita et al. 2000; Gupta et al. 2005; Zhu et al. 2008). Such core collections of reduced size are extremely useful for sequence polymorphism mining and for associating these polymorphisms with phenotypic traits. Association mapping or linkage disequilibrium (LD) mapping provide the advantage of tracking genetic polymorphisms that control phenotype variations among large panels of accessions, giving access to multiple alleles and thus increasing the efficiency of genetic resources exploitation. In these approaches, the main constraint relies on the genetic structure of the population and the size of the accession panel under investigation. Testing for statistical associations between the genotypes at the marker loci and the phenotypes in a sample of accessions is directly affected by the presence of groups of related accessions with different allele frequencies and may lead to false associations (Freedman et al. 2004). Then, the core-collection will be optimized if it maximizes the genetic diversity found in the whole collection and spans the full range of phenotypic variation (Ranc et al. 2010, 2008). Thus, preliminary analyses of the structure of the genetic diversity within the whole collection of accessions have to be performed together with an evaluation of the range of phenotypic variation. This prerequisite will further allow the selection of core-collections for SNP mining and for association or LD mapping.
Pepper (Capsicum spp.) is a complex of species originating from the intertropical America. The Capsicum genus belongs to the Solanaceae family and includes 27 recognized species (Baral and Bosland 2002). The taxonomic structure of the genus was established from a multidisciplinary approach using numerical taxonomy, cross fertility and cytogenetics, biochemical, geographical and ethnobotanical data (Pickersgill et al. 1979; Pickersgill 1991) providing evidence for 5 distinct cultivated species: C. annuum L., C. frutescens L., C. chinense Jacq., C. baccatum L. and C. pubescens Ruiz et Pav., which originated from distinct domestication events and primary diversification centers, where related wild species still coexist. Based on cross fertility and cytogenetics, the cultivated and the wild species were grouped into 3 genetic pools. C. annuum, C. frutescens and C. chinense form the first genetic pool (also named the white flowered species) which was related to the wild progenitor C. annuum var. glabriusculum (Dunal) Heiser et Pickersgill, C. baccatum and the wild relative C. baccatum var. baccatum Esbaugh (C. microcarpum Chodat et Hassl.) form the second genetic pool and C. pubescens together with the wild species C. eximium Hunz. and C. cardenasii Heiser et Smith form the 3rd genetic pool, those 2 pools being sexually isolated from each other and from the first one. Since the 1990s, genetic analyses using isozymes, nuclear and chloroplastic DNA markers confirmed this structure and increased our knowledge of the relationships between wild and domesticated species (Loaiza Figueroa et al. 1989; Rodriguez et al. 1999; Walsh and Hoot 2001; Toquica et al. 2003; Ince et al. 2010; Jeong et al. 2010; Ibiza et al. 2012). Genetic diversity was also explored in restricted geographic areas (mainly Mexico and Andean area) where cultivated species and wild relatives coexist, giving evidence for a genetic shift from wild to cultivated populations and a relative loss of genetic diversity in the cultivated environments, promoting rules for genetic resource conservation (Hernandez-Verdugo et al. 2001; Votava et al. 2002; Aguilar-Meléndez et al. 2009; Albrecht et al. 2012; González -Jara et al. 2012; Pacheco-Olvera et al. 2012).
Since its domestication in pre-columbian times, peppers have migrated worldwide. Evidence shows that it was firstly introduced from the West Indies into Europe in March 1,493, with the first travel of Christopher Columbus (Somos 1984; Andrews 1984). Successive migrations further occurred from the east coast of Central and South Americas through the Atlantic Ocean to Europe and Africa during the XVIth and XVIIth centuries, but also from the West coast of Peru through the Pacific Ocean to South-East Asia during the XVIIth and XVIIIth centuries. Trade routes between Europe, Middle-East and Asia promoted additional introductions and reciprocal exchanges, so that multiple introductions were rapidly cultivated in most tropical, mediterranean and temperate regions of the world. In these secondary diversification centers, thousands of landraces have been selected for 4–5 centuries by growers to fit new environments and local consumption habits and trade, resulting in the awesome phenotypic diversity of pepper cultivars (Nuez et al. 1996; Bosland and Votava 2000; Djian-Caporalino et al. 2007). C. annuum was the most successful in this conquest, although C. chinense and C. frutescens became also popular in Africa and Asia, whereas C. baccatum and C. pubescens mostly remained in South America and Andean regions. Genetic distances between cultivars or landraces was mainly explored within C. annuum and always among restricted sets of accessions (10 < n < 200) (Prince et al. 1992; Lefebvre et al. 1993; Paran et al. 1998; Lefebvre et al. 2001; Tam et al. 2009; Jung et al. 2010; Moses and Umaharan 2012; Nicolaï et al. 2012). These studies showed that DNA polymorphism rate is rather constant within species whatever the markers used and generally higher than the polymorphism observed in other autogamous solanaceae like tomato (Solanum lycopersicum L.), allowing intraspecific genetic mapping and cultivar identification. Structure of the genetic diversity within species was rarely analysed, except by Moses and Umaharan (2012) who showed relationships between the phylogenetic clusters and geographic distribution of C. chinense, and Lefebvre et al. (2001) and Tam et al. (2009) who revealed the narrow genetic basis of sweet and large fruited C. annuum cultivars compare to exotic landraces and the partial match between distances based on morphological traits and markers alleles. The molecular characterization of a larger panel of Capsicum genotypes, providing a more complete view of the differentiation between pepper cultivars and landraces worldwide is expected to provide a better understanding of the relationships between landraces and to enable us to establish core-collections for further studies of the impact of selection on genetic diversity.
With this aim, we genotyped the INRA Capsicum collection which includes 1,352 non redundant accessions from 89 different countries, with a large majority of C. annuum landraces, but also representatives of 10 additional cultivated or wild species (Sage-Palloix et al. 2007), using 28 SSR loci spanning 11 of the 12 pepper chromosomes. Model-based analysis structured this collection into 6 clusters, including 3 distinct clusters for C. annuum, which were related to large cultivar types differing in plant and fruit traits as a result of selection. These data were used to establish core collections with different sizes for further SNP mining or genetic association studies.
Materials and methods
Pepper germplasm collection
The pepper (Capsicum spp.) germplasm collection maintained at INRA, Unité de Génétique et Amélioration des Fruits et Légumes includes 1,352 non redundant accessions from 11 Capsicum species which were collected since 1959 from 89 distinct countries mostly from European (~35 %), American (~31 %), Asian (~22 %) and African (~9 %) continents (Sage-Palloix et al. 2007). Capsicum accessions are mostly landraces from the cultivated species: C. annuum (1,063 accessions, including 27 wild types (C. annuum var. glabriusculum)), 92 C. chinense, 51 C. frutescens, 107 C. baccatum, 18 C. pubescens and representatives of 6 wild species: 13 C. chacoense Hunz., 3 C. cardenasii, 2 C. eximium, 1 C. galapagoense Hunz., 1 C. microcarpum (C. baccatum var. baccatum) and 1 C. praetermissum Heiser et Smith. These accessions are maintained and multiplied by strictly controlled selfing in insect-proof greenhouses (except the wild allogamous C. eximium and C. cardenasii accessions where plants from the same accession are intermatted). These accessions (supplementary material) are registered in an internal data base (also available in the European Solanaceae Network: http://www.ecpgr.cgiar.org/germplasm_databases/list_of_germplasm_databases/crop_databases/crop_database_windows/pepper.html).
Phenotypic trait measurements
All the accessions were evaluated for 21 plant and fruit descriptors in relation with IPGRI descriptors for Capsicum (IPGRI, AVRDC and CATIE 1995) and resistance to several pathogens (Sage-Palloix et al. 2007). The plant and fruit traits were measured from two repeats of 3 individual plants during 2 years in a row, as described in Sage-Palloix et al. (2007) and in Barchi et al. (2009). In the present work, we used 3 primary plant traits: the flowering earliness is the time between sowing and first flower anthesis (in days, relative to the standard ‘Yolo Wonder’), the length of the primary axis is the length between cotyledon node and first fork (in cm), the number of leaves on the primary axis, and 4 fruit traits : the fruit length (Frl in cm), the fruit diameter is the maximum width generally at the proximal part of the fruit (Frd in cm), the apical fruit width (in cm) is the diameter measured at a distance of 5 % of the fruit length from the apical end (for example, in a 10 cm long fruit, the diameter is measured 0.5 cm (0.05 × 10) from the apical end), and the pericarp thickness (in mm). The ratio between fruit length and width (Frl/Frd) was also calculated. These traits are under polygenic inheritance and governed by QTLs spanning the whole genome. In the collection as well as segregating progenies, fruit traits were shown to be uncorrelated with plant traits, but positive correlations were significant between plant traits (ρpearson from 0.26 to 0.5) and between Frd and pericarp thickness (ρpearson = 0.6) (Sage-Palloix et al. 2007; Barchi et al. 2009; Alimi et al. 2013).
DNA extraction and microsatellite genotyping
DNA was extracted from pools of 6 young plantlets (fresh leaf tissue 3 weeks after sowing) per accession as described by Fulton et al. (1995). The DNA was resuspended in 100 μl of Tris EDTA solution and quantified with Nanodrop system and a picogreen assay (Invitrogen) according to manufacturer’s protocol. A set of 28 microsatellite markers publicly available from Lee et al. (2004); Yi et al. (2006); Nagy et al. (2007) and Portis et al. (2007) was chosen on the basis of their distribution on the genetic map, spanning 11 of the 12 pepper chromosomes (Table 1). PCR amplifications were performed in a 10 μl reaction volume containing 25 ng of genomic DNA as template. Forward primers were 5′-end labelled with FAM, VIC, or NED for analysis on an Applied Biosystems 3730xI DNA Analyzer on the “Gentyane” Platform (INRA Clermont Ferrand). GeneMapper 3.7 software (Applied Biosystems) was used to evaluate the size of the alleles.
Genetic diversity analysis
The number of alleles, the number of genotypes, the Nei’s unbiased gene diversity index (He), the observed heterozygosity (Ho), and Polymorphism Information Content (PIC) were calculated using the Power Marker version 3.25 software (Liu and Muse 2005, http://www.powermarker.net). The linkage disequilibrium (LD) was expressed by r 2 values and the statistical significance (P value) of the observed LD was estimated by Monte-Carlo approximation of Fisher’s exact test, with 1,000 permutations.
Structure of the collection
To infer the population structure of the pepper collection, we used the model-based clustering algorithm implemented in the computer program Structure version 2.3.3 (Pritchard et al. 2000). This algorithm uses a multilocus genotype to identify a predetermined number (K) of clusters that have distinct allele frequencies and assigns portions of individual genomes to these clusters. It proceeds by assuming that observations are randomly drawn from a parametric model and inference for the parameters allows estimation of ancestry probability from each putative cluster, for all individuals. Since pepper accessions used are highly homozygous (autogamy plus self-pollination of accessions), we used a haploid setting and the heterozygous loci were changed in missing data. We used the admixture model assuming correlation among allele frequencies. Ten runs were taken into account for each value of K (K is the number of clusters to be inferred), for K ranging from 1 to 10. In each run, we used a burn-period of 500,000 Markov Chain Monte Carlo iterations and then 250,000 iterations for estimating the parameters. Pr(X|K) (i.e. the posterior probability of the data (X) given K) and the associated standard deviation was computed for each simulation, and the optimal K value (Kopt) was inferred from the formula established by Evanno et al. (2005). For each Kopt, individuals were assigned into a cluster when their proportion of membership into this cluster was higher than 50 % (supplementary material).
Neighbour-joining tree and principal coordinate analyses
Genetic distance matrices between pairs of accessions were estimated from an index of dissimilarity based on the simple matching method for SSR alleles, and the standardized Euclidean distances for quantitative phenotypic traits. The graphical representation of the neighbour joining trees and principal coordinate analyses were performed with the DARwin 5.0.158 software (Perrier and Jacquemoud-Collet 2006).
Core collection sampling
For sampling core collections, we used the Maximization (M) algorithm implemented in MSTRAT software version 4.1 (Gouesnard et al. 2001) which permits to maximize the number of alleles captured in the sample (allelic richness) and compared the result to a random sampling strategy. The minimum number of accessions in each core collection to capture all alleles present in the whole collection was evaluated by sampling simulations in this collection. The core collections were built using all SSR data alone (nested core collection) or together with phenotypic alleles for 3 plant phenotypic traits (flowering earliness, number of leave, primary axis length), and 3 fruit traits (fruit length, fruit diameter and pericarp thickness). For phenotypic alleles, quantitative phenotypic data were split into 10 classes of equal amplitudes. For evaluation of core collection’s minimal size and for accessions sampling, 20 replicates with 30 iterations per replicate were performed.
Results
Microsatellite diversity across the Capsicum species
The diversity pattern of the 28 SSR loci across the 1,352 accessions of the whole Capsicum collection (Table 1) revealed a highly variable number of alleles, ranging from 3 to 47 distinct alleles per locus with an average of 18.21. The Nei’s unbiased gene diversity indices (He) as well as the PIC which both represent the number of alleles and their distribution remained generally high (average of 0.7 and 0.67 respectively). The observed heterozygosity was very low (<0.085), as expected from the plant accessions maintained and multiplied through selfing. A few markers did not deliver any amplicon in some species (null alleles): particularly in C. pubescens, C. cardenasii nor C. eximium (Epms426, HpmsE064 and Gpms100), C. baccatum (HpmsE051 and EPMS342) and C. chacoense (HpmsE051). The 1,352 Capsicum accessions were previously screened to remove redundant accessions according to passport and phenotypic descriptors. However, for the 28 SSR loci, some accessions displayed strictly the same multilocus genotypes. These genetically redundant accessions were removed from the subsequent analyses together with accessions delivering more than 4 missing data (over 28 loci), leading to a final panel of 1,210 accessions including 908 cultivated C. annuum (vs. 1036), 104 C. baccatum (vs. 107), 87 C. chinense (vs. 92), 48 C. frutescens (vs. 51) and 10 C. chacoense (vs. 13). The accessions remained unchanged in the other species (Table 2).
The mean SSR diversity indices were considered in the different Capsicum species including at least 10 accessions (Table 2). The mean number of alleles for each species (2.68–12.57) is biased due to the unbalanced number of accessions between species. Thus, it is more relevant to consider the He and PIC indices that are maximum in the C. annuum var. glabriusculum sub-species with values of 0.78 and 0.75, and lower but rather similar between the other species (0.47–0.59 for He and 0.44–0.54 for PIC). The high homozygosity of accessions in every species, expected from the rejuvenation through strictly controlled selfing, is confirmed, with the highest heterozygous frequency in the wild C. chacoense (8.6 %).
Genetic structure of the collection
Only two pairs of markers displayed weakly significant linkage disequilibrium: Gpms 101-Gpms29 with r 2 = 0.24; and HpmsE051-Hpms2-24 with r 2 = 0.20. These two pairs of markers were linked respectively at 3.2 and 3.0 cM (Kosambi) on the pepper genetic map. Because of these low r 2 values and independence of all the other loci, the 28 SSR markers were used for the Structure analysis.
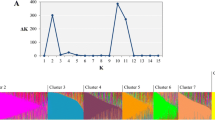
After the Evanno et al. (2005) correction, the genetic structure in the complete collection (1,210 accessions) displayed 6 clusters (K = 6, Fig. 1). The clusters 1, 2 and 3 displayed some admixture, with accessions partly belonging to 2 or 3 of these clusters, based on their genome proportion attributed to each cluster. These first 3 clusters included all the cultivated C. annuum accessions. The clusters 4, 5 and 6 displayed a clear cut structure with no or very few admixture. The 4th cluster included all the C. frutescens and C. chinense accessions, together with the single C. galapagoense. The 5th cluster included all the C. baccatum accessions, together with the single C. baccatum var. baccatum accession. The 6th cluster included all the C. pubescens accessions, together with the accessions of the wild species C. eximium, C. cardenasii, C. praetermissum, and also the 10 C. chacoense accessions. This model based clustering closely corresponded to the known taxonomic groups of the Capsicum genus, except for the C. annuum var. glabriusculum accessions which were distributed in several clusters. Among the 27 accessions classified as wild C. annuum var. glabriusculum, 9 were included into cluster 1 (C. annuum), 14 into the cluster 6 including 3 accessions with an admixed genome between clusters 6 and 1 or 4, and 4 accessions located into the cluster 4 (C. frutescens and C. chinense) but with an admixed genome with cluster 6 (3 accessions) or 1 (one accession).

Model-based populations in the whole Capsicum collection based on allelic variants at the 28 SSR loci. Upper graph: determination of K optimal following the method of Evanno et al. (2005). Lower graph: classification of the 1,210 Capsicum individuals using Structure 2.3.3. The distribution of the individuals into distinct clusters by the model-based method is indicated by the color code in the legend box. Capsicum species included in each cluster are indicated
A phylogenetic tree for the whole collection, based on Nei’s genetic pairwise distances was constructed using UPGMA procedure as implemented in the DARwin 5.0 software. This tree generally confirms the previous model based clustering, and brings more precisions in agreement with the taxonomic classification of Capsicum species (Fig. 2). Indeed, the C. chacoense accessions are clearly separated from the C. pubescens accessions, similarly, the C. chinense accessions are clearly in a distinct branching than the C. frutescens accessions. Finally, the wild C. annuum var glabriusculum were distributed in different branches: at the root of and within the C. annuum branches (13 accessions) or at the root of the C. frutescens and C. chinense branches (12 accessions), or close to the C. chacoense group (2 accessions). The dispersion of the previous cluster 6 into distinct branches of the tree also suggests that it included different genetic groups with high allele diversity but poor representation (less that 15 accessions per species). Finally, the cultivated C. annuum accessions are distributed in several branches in the lowest half of the tree with a large group corresponding to the previous cluster 1, but displayed a slightly more complex pattern for the previous clusters 2 and 3 which are subdivided into a few subgroups.

Phylogenetic tree showing the genetic diversity of the pepper germplasm collection. The tree was produced using the neighbor-joining UPGMA method based on the 28 SSR markers. The colors (and small numbers representing accessions) correspond to the previous clusters from the model-based analysis as in Fig. 1. Capsicum species of the accessions are indicated (in bold :cultivated species, normal font: wild species) and correspond to the main phylogenetic branches
Genetic and phenotypic diversity of the three clusters of C. annuum
The model based analysis with the Structure software delivered 3 distinct clusters within the C. annuum accessions (Fig. 1) with more admixed accessions than the previous clusters: in the cluster 1, 22 % of accessions (72/326) had between 80 and 50 % of membership into this cluster, in cluster 2 this proportion was 21 % (40/201) and in cluster 3: 17 % (64/386). These 3 C. annuum clusters differ in their genetic diversity, with a higher He value for the cluster 1 (He 0.64) than for clusters 2 and 3 (He 0.44 and 0.40 respectively) (Table 3). In a first attempt to explore the diversity in phenotypes and origin of these distinct clusters, a phenotypic characterization was attempted by comparing the means for plant and fruit parameters used in pepper germplasm description. The 3 clusters were clearly distinct with significantly different average values for most plant and fruit descriptors (Figs. 3 and 4).

Average values of the 3 cultivated C. annuum clusters defined by the model-based analysis for 8 plant and fruit traits. Vertical bars represent the 95 % confidence interval. Flowering earliness expressed in days relative to the Yolo Wonder control, primary axis length, fruit length, fruit diameter and fruit apical width in cm, fruit pericarp thickness in mm

Distribution of the distinct fruit types in the three C. annuum clusters determined from the model based analysis. Fruit types were defined in Sage-Palloix et al. (2007)
The cluster 1 was characterized by late flowering plants (+3 days in average), with a long primary axis (28 cm) developing at least 14 leaves before flowering. Fruits from these accessions were small in length and particularly in diameter (1.9 cm in average) resulting in an elongated shape (4.7 times longer than large), with a pointed blossom end (very small apical width), and a thin pericarp (1.5 mm). That is characteristic for most small and elongated fruited peppers which represent 82 % of the accessions from this cluster (Fig. 4). This cluster was highly diversified in the geographic origins of the accessions, including traditional Mexican cultivars (‘Pasilla’, ‘Anaheim’, ‘Serrano’ types and ‘Criollo de Morelos’), from Asia (‘Perennial’ from India, ‘Xian jiao’ types from China) and from Africa (‘H3’ from Ethiopia, ‘Chatah’ from Sudan). These late flowering and small-fruited cultivars mostly originate from subtropical areas but the cluster also includes many cultivars that became traditional in temperate and Mediterranean countries like ‘Espelette’ pepper from France which appeared close to the Mexican ‘Pasilla Apaseo’.
The cluster 2 is characterized by early flowering plants (−3.5 days in average) with shorter primary axis (22 cm) bearing a lower number of leaves (9.5). The fruits are longer and larger than the previous cluster, resulting in a 2.8 Frl/Frd ratio with an obtuse apical end and a much thicker pericarp (4.2 mm). This corresponds to the triangular and horn shaped peppers but also to elongated and large peppers, which represent 40 and 35 % of the accessions respectively (Fig. 4). Considering the geographic origin, this cluster displayed a clear predominance of central European origin. Indeed, 147 of the 201 accessions from cluster 2 (73 %) are local cultivars originating from central Europe (Hungary, former Yugoslavia and Czechoslovakia, Romania, Poland, South Russia, Bulgaria) whereas these countries represent only 18 % of the geographical origins of the whole C. annuum collection. This cluster can be characterized by the traditional cultivars with elongated fruits like ‘Hatvani’, conical fruits with light green or ivory immature color like ‘Podarok Moldavia’, ‘Feherozon’, ‘Cecei’, but also a few blocky fruits with ivory color like ‘Bela Krupna’ or ‘Paradicsom’ from Yugoslavia and Hungary. Another characteristic of this cluster is the presence of the traditional Turkish cultivars with horn or conical shaped fruits (10 landraces from the ‘Sivri’, ‘Carliston’ or ‘Maras’ types).
The cluster 3 included plants with intermediate earliness but axis growth and development close to the cluster 2. Fruits are close to the cluster 2 in length but significantly larger (6 cm) resulting in an average Frl/Frd ratio of 1.8. The mean apical end is much more large and lobate and the pericarp is thick (4.8 mm). This clearly corresponds to the large fruited peppers with blocky or rectangular shape which contribute to 61 % of the accessions of this cluster (Fig. 4). Geographic origins in this cluster are diversified, including the traditional cultivars with very large (up to 600 g) and rectangular fruits from Mediterranean Europe (‘Largo de Reus’ and ‘Largo Valenciano’ from Spain, ‘Lagne’ and ‘Lamu’ from France), the large blocky fruits from Italy (‘Quadrato Asti’), smaller blocky fruits from USA (‘Yolo’, ‘California Wonder’), from Netherlands (‘Mavras’), Poland (‘Oda’), China (‘Zao Feng’, ‘Ben Xi’). In this cluster were also located several accessions with thick pericarp but triangular, tomato, cherry or heart shaped fruits like ‘Fresno’ or ‘Cherry bomb’ from USA, ‘Morron Conserva’ or ‘Niora’ from Spain. These cultivars present an admixed genome between the cluster 3 and 2 or 1 and located in the isolated branch of the phylogenetic tree (Fig. 2), between the 2 branches corresponding to the cluster 2.
Construction of core collections
Sub-samples of 8, 16, 32, 64, and 128 accessions of C. annuum were selected, based on their genotypes at the 28 SSR loci using the Maximization (M) strategy algorithm implemented in MStrat software v.4.1. In this strategy, the accessions from the smaller samples were included in the successive larger samples (nested core-collections) (supplementary material). These successive core collections captured 37, 55, 71, 85, and 89 % of the alleles from the whole C. annuum collection (Table 4). In the largest core collection, the 128 accessions are distributed in the 3 different C. annuum clusters with a prevalence of cluster 1 that corresponds to the higher diversity of this cluster. However, decreasing the size of the core collections strongly affect this distribution, so that the accessions from clusters 3 and 2 were underrepresented or lost in the smaller core collections. The M strategy algorithm, using alleles at SSR loci, permits to select the smallest samples while maximizing the genetic diversity which maybe favourable for sequence diversity analyses and SNP mining. However the deficit in accessions from the cluster 3 (large fruited cultivars) and cluster 2 (early flowering plants and conical or long fruited cultivars) affects the representativeness of the smallest as well as large samples for horticultural traits, which may not be favourable for association analyses with these traits.
A larger core collection of C. annuum was built with the objective to optimize the contribution of the C. annuum clusters, and to maximize both the genetic and phenotypic diversity. This was achieved by selecting separate core-collections from each of the 3 C. annuum clusters which size was expected to be proportional to the gene diversity in each cluster (He = 0.64, 0.44 and 0.40 for clusters 1, 2 and 3, Table 3). Moreover, the sampling of accessions using the M-algorithm was performed on the basis of their alleles at the 28 SSR markers and of their phenotypic alleles for the 6 primary traits (Flowering date, axis length, number of leaves, fruit length, fruit width, pericarp thickness). Random and M sampling strategies were compared for the allelic richness captured when sampling a core collection of n individuals. The difference between the random and M curves (Fig. 5) clearly shows that the M strategy performed better in sampling the core collections from the 3 C. annuum clusters. The plateau of the M curve was reached at 163 accessions in cluster 1, 100 in cluster 2 and 193 in cluster 3 although showing a rather flat curve and poor score gain above n = 90. The contribution of each cluster to the final core collection was adjusted according to their respective gene diversity as written above, resulting in 142 accessions from cluster 1, 97 from cluster 2, and 93 from cluster 3 with accessions which were the most frequently sampled in the 20 replicates for each cluster. This final core collection of 332 accessions (supplementary material) captured 97 % of the SSR as well as phenotypic alleles. Compared to the whole C. annuum collection (908 accessions), 332 SSR alleles were captured (instead of 352) with a high gene diversity: He = 0.633, instead of 0.591 in the whole C. annuum collection which also results from the smaller population size. The principal coordinate analyses of the whole C. annuum collection and of the core collection were performed for the genotypic and the phenotypic data (Fig. 6). The areas of distribution of accessions across the first PCoA plan were not modified in the core collection except for the lower density, attesting the representativeness of the selected sample.

Comparison of efficiency between random and maximization (M) sampling strategies in the Capsicum annuum clusters defined from Structure analysis. Score, which represents allelic richness, is plotted against the size of the core collections. The efficiency of the M strategy is represented by the blue continuous line and the random strategy is represented by the red dotted line

Principal coordinate analysis of the C. annuum collection and the large core collection of 332 accessions. A and B: analyses based on the alleles at the 28 SSR loci with dissimilarity matrices built according to the simple matching coefficient. C and D: analyses based on the quantitative fruit and plant traits with dissimilarity matrices built according to the standardized Euclidean distances. Graphs A and C represent the whole C. annuum collection (908 accessions) and graphs B and D the core collection sampled using SSR and phenotypic traits (332 accessions). Colors according to the Structure 2.3.3 clustering information: blue cluster 1, red cluster 2, green cluster 3
Discussion
The Capsicum collection analysed in this report resulted from nearly 50 years of collection and exchanges (1962–2010) with pepper geneticists worldwide. It does not result from an exhaustive or a balanced sampling plan of genotypes in a given area, but the 1,352 accessions collected from 89 countries were maintained and selected to avoid redundancy and maximize diversity between accessions based on phenotypic traits and geographic origins. This large collection was fingerprinted using 28 highly polymorphic SSR loci distributed overall the pepper genome, and the data set delivered new global overview on the structure of genetic diversity among the major species of the Capsicum genus and within the C. annuum germplasm.
At the genus level, the model based structure analysis, completed with the neighbour joining analysis of the whole Capsicum collection clearly separates the known Capsicum taxonomic groups and species. However, we were surprised by the discrepancy between the results from these two analyses. The model based analysis clustered together accessions from different species, particularly in clusters 4 (C. chinense and C. frutescens) and 6 (18 C. pubescens, 10 C. chacoense and 14 C. annuum var. glabriusculum), whereas the neighbour joining analysis separated these species. Particularly, among the 14 C. annuum var. glabriusculum, 12 were relocated between the branches of the white flowered species in the neighbour-joining tree, which is more coherent with previous knowledge. It is highly probable that the model based analysis grouped together these taxonomic groups which were represented by a weak number of accessions and were genetically diversified and distant from the other groups. Considering the two approaches, all the accessions were clearly distributed in the C. baccatum group or the C. pubescens group, both from South-America, or one of the 3 species of the white flowered group including C. chinense, C. frutescens and C. annuum, the later from Central and North America. C. chacoense, which was previously classified in the white flowered species group due to its morphotype and partial sexual compatibility with C. annuum (Pickersgill 1991) grouped between the South American species C. baccatum and C. pubescens in our phylogenetic tree, according to its origin (Bolivia-Paraguay) and in agreement with chloroplast DNA analysis of Walsh and Hoot (2001) and the nuclear SNP analyses of Jeong et al. (2010).
Less was known from the wild accessions of C. annuum (var. glabriusculum) and its relationships with the cultivated species. Very tiny ovoid fruits (less than 1 g), deciduous with erect habit, and bushy plants characterize these accessions. They appeared highly diversified for SSR alleles despite their weak representation in our collection (27 accessions). Most of these accessions were introduced from B. Pickersgill in 1976 and 1977 (var. aviculare, Pickersgill et al. 1979). C. annuum var. glabriusculum has been commonly cited under the name “Chiltepin” and considered as the wild parent of the cultivated C. annuum (Bosland and Votava 2000; Votava et al. 2002; Aguilar-Meléndez et al. 2009; González-Jara et al. 2012). However, the former numerical taxonomic studies (Pickergill et al. 1979) showed this wild form to relate equiprobably to cultivated white flowered species C. annuum, C. frutescens and C. chinense, suggesting a common wild ancestor to these species. Further karyotype analyses (Pickersgill 1991; Moscone et al. 2007) demonstrated distinct karyotypes in C. annuum var. glabriusculum, with one or two pairs of (sub) telomeric chromosomes which are specific of C. frutescens and C. chinense or of the domesticated C. annuum respectively. In the neighbour-joining analysis, the C. annuum var. glabriusculum accessions distributed in several clusters, with one major group of 13 accessions closely related to C. annuum and 12 accessions closely related to C. frutescens (Fig. 2). The 13 C. annuum related accessions originated from Mexico (including Chiapas, Oaxaca, Vera-Cruz and Nuevo Leon states), from Florida (2 accessions) and Texas (1 accession). Among the 12 C. frutescens related accessions only 3 were collected in Mexico and 9 in Central and South America including Guatemala, Nicaragua, Costa-Rica, Panama, and Ecuador. Interestingly, this validates and refines their positions relatively to the cultivated species from the white flowered group. It strongly suggests distinguishing the wild relatives of C. annuum with two subtelomeric chromosome pairs which originated from Mexico and North America, from the wild relatives of C. frutescens and/or C. chinense with one subtelomeric chromosome pair, mostly originating from Central and South Americas. The two accessions from Colombia which cluster close to the C. chacoense group may have retained some shared (ancestral) alleles under the hypothesis that Capsicum initially originated from South America. Endly the C. galapagoense accession which was never subjected to genome analysis clearly grouped together with the Central American and white flowered accessions.
Considering the cultivated C. annuum, the model based analysis clustered the 908 accessions into 3 main genetic clusters. These clusters mostly corresponded to cultivar types with a clear-cut separation between small and elongated fruited peppers also characterized by their thin pericarp and late flowering tall plants (cluster 1) in opposition to the large fruited and fleshy (thick pericarp) peppers (cluster 3) which displayed a lower genetic diversity. An intermediary cluster (cluster 2) included the conical and elongated fruited peppers with a rather thick pericarp, also characterized by their earliness and shorter stems (compact plants, some of them with determinate growth). In Tam et al. (2009), a similar structure was observed with 64 C. annuum accessions from the same collection, using SSAP markers. Their two clusters of sweet and large fruited peppers roughly corresponded to our clusters 2 and 3, despite pungent peppers appear more largely present in our cluster 2, probably due to a much larger sampling. The clear origin of the cluster 2 (Central Europe and Turkish cultivars) also contribute to identify this cluster as a distinct genetic pool. Their early flowering and short stem (compact habit) traits also indicate an adaptation to short production cycle (short summer) in continental climates of central Europe, in opposition to the late flowering and tall plants of the cluster 1 which better reflects the adaptation to intertropical climates. The neighbour joining tree roughly confirmed this structure with 3 clusters, although the accessions from the cluster 2 split into two main branches (Fig. 2) separated by a branch with accessions from cluster 3. These accessions are pepper landraces with thick pericarp but smaller fruits (heart, tomato, or cherry peppers), often used for processing. Most of these intermediate branches include accessions with admixed genome. In the neighbour joining analysis, the splitting of accessions from cluster 2 into different branches, together with their intermediary fruit traits does not argue in favour of a distinct genetic origin as hypothesized by Tam et al. (2009). These cultivar groups more probably represent an intermediate genetic pool in the evolution from small to large fruited cultivars, or recombinants between the extreme small fruited cultivars from cluster 1 and large fruited cultivars from cluster 3. In every case, the genetic structure related to the cultivar types or cultigroups as shown in many vegetables like Brassica oleracea L. with the brussel sprout, cabbage, cauliflower and broccoli groups (Louarn et al. 2007), in lettuce with the butter head, crisp head, roman or iceberg types (Simko 2009), and also in zucchini (Ferriol et al. 2003), cucumber (Jing et al. 2012) or eggplant (Hurtado et al. 2012) cultivar types. It results from the history of consumer driven selection for fruits and plant traits in the primary and secondary diversification centers.
The structure by geographic origins of the landraces was weakly visible when considering the whole C. annuum collection. Contrarily to previous genetic analyses of pepper collections from restricted origins (Aguilar-Meléndez et al. 2009; González-Jara et al. 2012; Albrecht et al. 2012) which generally fitted the geographic structure, we examined C. annuum accessions from many (89) countries. Only the accessions from cluster 2 clearly originated from Turkey and central Europe indicating that these landraces are related, probably resulting from a foundation effect followed by local selection and relative confinement to this production area. The two other clusters included landraces and cultivars from highly diversified geographic origins. A more detailed analysis within each cultivar type can reveal groups of accessions with shared geographic origin. However, most of these groups also include cultivars which were collected in exotic countries. When considering such a large panel, genetic differentiation between cultivar types interferes or tends to dominate the differentiation between geographic origins. This attests the numerous and complex migration events of many pepper genotypes resulting from human migrations, their adoption in a new country and their further local selection.
A core collection represents the genetic diversity of a crop with a minimal redundancy in order to reduce the costs or facilitate maintenance and/or evaluation efforts. However, such subsamples have to be optimized according to their further exploitation (Marita et al. 2000; Ranc et al. 2008). In pepper, only one core collection has been already published (Zwendie et al. 2004), based on phenotypic traits and cluster analysis. We established several core collections based on SSR alleles in order to maximize the genetic diversity but also including phenotypic traits to optimize further screening and analyses of horticultural traits. The nested core collections ranging from 8 to 128 accessions represent from 37 to 90 % of the genetic diversity of C. annuum and its wild relatives (var. glabriusculum). The smallest samples are adequate for genome sequencing and SNP mining (Nicolaï et al. 2012) since pepper possess a large genome of 3.3 Gb and genome sequencing remains expensive. However these core collections do not span the full range of phenotypic variability, particularly in pepper cultivars from cluster 2 and 3 which are of high interest to look for association between gene polymorphisms and horticultural traits including plant growth and fruit size and shape. The large sample of 332 accessions with 97 % of genetic diversity provides a complete sample for the representation of horticultural types and is expected to provide a more balanced frequency of alleles retained by long-term selection and determining horticultural traits in C. annuum.
References
Aguilar-Meléndez A, Morrell PL, Roose ML, Kim SC (2009) Genetic diversity and structure in semiwild and domesticated chili peppers (Capsicum annuum; Solanaceae) from Mexico. Am J Bot 96:1190–1202
Albrecht E, Zhang D, Saftner RA, Stommel JR (2012) Genetic diversity and population structure of Capsicum baccatum genetic resources. Genet Resour Crop Evol 59:517–538
Alimi NA, Bink MCAM, Dieleman JA, Nicolaï M, Wubs M, Heuvelink E, Magan JJ, Voorrips RE, Jansen J, Canas-Rodrigues P, Vercauteren A, Vuylsteke M, Song Y, Glasbey C, Barocsi A, Lefebvre V, Palloix A, van Eeuwijk FA (2013) Genetic and QTL analyses of yield and a set of physiological traits in pepper. Euphytica 190:181–201
Andrews J (1984) Pepper. University of Texas Press, Austin, Texas, The domesticated Capsicum. 163 p
Baral JB, Bosland P (2002) An updated synthesis of the Capsicum genus. Capsicum and Eggplant Newsl 21:11–21
Barchi L, Lefebvre V, Sage-Palloix AM, Lanterri S, Palloix A (2009) QTL analysis of plant development and fruit traits in pepper using selective phenotyping. Theor Appl Genet 118:1157–1171
Bosland PW, Votava EJ (2000) Peppers: vegetable and spice Capsicum. CABI Publishing, Oxon
Djian-Caporalino C, Lefebvre V, Sage-Daubèze AM, Palloix A (2007) Capsicum. In: Singh RJ (ed) Genetic resources, chromosome engineering and crop improvement series, Vol 3, Vegetable crops. CRC Press, Florida (USA), pp 185–243
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
FAO (2010) The second report on the state of the world’s plant genetic resources for food and agriculture. Rome, 370 p. http://www.fao.org/docrep/013/i1500e/i1500e00.htm
Ferriol M, Picό B, Nuez F (2003) Genetic diversity of a germplasm collection of Cucurbita pepo using SRAP and AFLP markers. Theor Appl Genet 107:271–282
Freedman ML, Reich D, Penney KL et al (2004) Assessing the impact of population stratification on genetic association studies. Nat Genet 36:388–393
Fulton TM, Chunwongse J, Tanksley SD (1995) Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol Rep 13:207–209
González-Jaral P, Moreno-Letelier A, Fraile A, Pinero D, García-Arenal F (2012) Impact of human management on the genetic variation of wild pepper Capsicum annuum var. glabriusculum. PLoS ONE 6(12):e28715. doi:10.1371/journal.pone.0028715
Gouesnard B, Bataillon TM, Decoux G, Rozale C, Schoen DJ, David JL (2001) MSTRAT: an Algorithm for building germplasm core collections by maximizing allelic or phenotypic richness. J Hered 92:93–94
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57:461–485
Hammer K, Arrowsmith N, Gladis T (2003) Agrobiodiversity with emphasis on plant genetic resources. Naturwissenschaften 90(6):241–250. doi:10.1007/s00114-003-0433-4
Hernandez-Verdugo S, Luna-Reyes R, Oyama K (2001) Genetic structure and differentiation of wild and domesticated populations of Capsicum annuum (Solanaceae) from Mexico. Plant Syst Evol 226:129–142
Hurtado M, Vilanova S, Plazas M, Gramazio P, Fonseka HH, Fonseka R, Prohens J (2012) Diversity and relationships of eggplants from three geographically distant secondary centers of diversity. PLoS ONE e41748
Ibiza VP, Blanca J, Cañizares J, Nuez F (2012) Taxonomy and genetic diversity of domesticated Capsicum species in the Andean region. Genet Resour Crop Evol 59:1077–1088
Ince AG, Karaca M, Onus AN (2010) Genetic relationships within and between Capsicum species. Biochem Genet 48:83–95
IPGRI, AVRDC, CATIE (1995) Descriptors for Capsicum (Capsicum spp.). International Plant Genetic Resources Institute, Rome, Italy
Jeong HJ, Jo YD, Park SW, Kang BC (2010) Identification of Capsicum species using SNP markers based on high resolution melting analysis. Genome 53:1029–1040
Jing LV, Qi J, Shi Q, Shen D, Zhang S, Shao G, Li H, Sun Z, Weng Y, Shang Y, Gu X, Li X, Zhu X, Zhang J, van Treuren R, van Dooijeweert W, Zhang Z, Huang S (2012) Genetic diversity and population structure of cucumber (Cucumis sativus L.). PLoS ONE 7:e46919
Jung JK, Park SW, Liu WY, Kang BC (2010) Discovery of single nucleotide polymorphism in Capsicum and SNP markers for cultivar identification. Euphytica 175:91–107
Lee JM, Nahm SH, Kim YM, Kim BD (2004) Characterization and molecular genetic mapping of microsatellite loci in pepper. Theor Appl Genet 108:619–627
Lefebvre V, Palloix A, Rives M (1993) Nuclear RFLP between pepper cultivars (Capsicum annuum L.). Euphytica 71:189–199
Lefebvre V, Goffinet B, Chauvet JC, Caromel B, Signoret P, Brand R, Palloix A (2001) Evaluation of genetic distances between pepper inbred lines for cultivar protection purposes: comparison of AFLP, RAPD and phenotypic data. Theor Appl Genet 102:741–750
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Loaiza-Figueroa F, Ritland K, Laborde Cancino JA, Tanksley SD (1989) Patterns of genetic variation of the genus Capsicum (Solanaceae) in Mexico. Pl Syst Evol 165:159–188
Louarn S, Torp AM, Holme IB, Andersen SB, Jensen BD (2007) Database derived microsatellite markers (SSRs) for cultivar differentiation in Brassica oleracea. Genet Resour Crop Evol 54:1717–1725
Marita JM, Rodriguez JM, Nienhuis J (2000) Development of an algorithm identifying maximally diverse core collections. Genet Resour Crop Evol 47:515–526
Moscone EA, Scaldaferro MA, Grabiele M, Cecchini NM, Garcia YS, Jarret R, Davina JR, Ducasse DA, Barboza GE, Ehrendorfer F (2007) The evolution of chili peppers (Capsicum—Solanaceae): a cytogenetic perspective. In: Proceedings of the VIth international Solanaceae conference. Solanaceae VI: Genomics Meets Biodiversity, Acta Horticulturae 745:137–169
Moses M, Umaharan P (2012) Genetic structure and phylogenetic relationships of Capsicum chinense. J Am Hort Sci 137:250–262
Nagy I, Stàgel A, Sasvári Z, Röder M, Ganal M (2007) Development, characterization, and transferability to other Solanaceae of microsatellite markers in pepper (Capsicum annuum L.). Genome 50:668–688
Nicolaï M, Pisani C, Bouchet JP, Vuylsteke M, Palloix A (2012) Discovery of a large set of SNP and SSR genetic markers by high-throughput sequencing of pepper (Capsicum annuum). Genet Mol Res 11:2295–2300
Nuez F, Gil R, Costa J (1996) El cultivo de pimientos, chiles y ajies. Mundi-Prensa, Madrid
Pacheco-Olvera A, Hernández-Verdugo S, Rocha-Ramírez V, González-Rodríguez A, Oyama K (2012) Genetic diversity and structure of pepper (Capsicum annuum L.) from Northwestern Mexico analyzed by microsatellite markers. Crop Sci 52:231–241
Paran I, Aftergoot E, Shifriss C (1998) Variation in Capsicum annuum revealed by RAPD and AFLP markers. Euphytica 99:167–173
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. http://darwin.cirad.fr/
Pickersgill B (1991) Cytogenetics and evolution of Capsicum L. In: Tsuchiya T, Gupta PK (eds) Chromosome engineering in plants: genetics, breeding, evolution. Elsevier, Amsterdam, pp 139–160
Pickersgill B, Heiser CB, McNeill J (1979) Numerical taxonomic studies of variation and domestication in some species of Capsicum. In: Hawkes JG (ed) The biology and taxonomy of the Solanaceae. Academic Press, New York, pp 679–699
Portis E, Nagy I, Sasvari Z, Stagel A, Barchi L, Lanteri S (2007) The design of Capsicum spp. SSR assays via analysis of in silico DNA sequence, and their potential utility for genetic mapping. Plant Sci 172:640–648
Prince JP, Loaiza-Figueroa F, Tanksley SD (1992) Restriction fragment length polymorphism and genetic distance among Mexican accessions of Capsicum. Genome 35:726–732
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181
Ranc N, Muños S, Santoni S, Causse M (2008) A clarified position for Solanum lycopersicum var. cerasiforme in the evolutionary history of tomatoes (Solanaceae). BMC Plant Biol 8:130. doi:10.1186/1471-2229-8-130
Ranc N, Muños S, Xu J, Le Paslier MC, Chauveau A, Bounon R, Rolland S, Bouchet JP, Brunel D, Causse M (2010) Genome-wide association mapping in tomato (Solanum lycopersicum) is possible using genome admixture of Solanum lycopersicum var. cerasiforme. Genes Genomes Genet 2:853–864
Rodriguez JM, Berke T, Engle L, Nienhuis J (1999) Variation among and within Capsicum species revealed by RAPD markers. Theor Appl Genet 99:147–156
Sage-Palloix AM, Jourdan F, Phaly T, Nemouchi G, Lefebvre V, Palloix A (2007) Structuring genetic diversity in pepper genetic resources: distribution of horticultural and resistance traits in the INRA pepper germplasm. In: Niemirowicz-Szczytt K (ed) Progress in research on Capsicum and eggplant. Warsaw University of Life Sciences Press, Warsaw, Poland, pp 33–42
Simko I (2009) Development of EST-SSR markers for the study of population structure in lettuce (Lactuca sativa L.). J Hered 100:256–262
Somos A (1984) The paprika. Akademiai Kiado, Budapest, ISBN 963 05 3299 9, p 302
Tam SM, Lefebvre V, Palloix A, Sage-Palloix AM, Grandbastien MA (2009) LTR-retrotransposons Tnt1 and T135 markers reveal genetic diversity and evolutionary relationships of domesticated peppers. Theor Appl Genet 119:973–989
Tang H, Sezen U, Paterson AH (2010) Domestication and plant genomes. Curr Opin Plant Biol 13:160–166
Toquica SP, Rodriguez F, Martinez E, Duque MC, Tohme J (2003) Molecular characterization by AFLPs of Capsicum germplasm from the Amazon department in Colombia. Genet Resour Crop Evol 50:639–647
Votava EJ, Nabhan GP, Bosland PW (2002) Genetic diversity and similarity revealed via molecular analysis among and within an in situ population and ex situ accessions of chiltepin (Capsicum annuum var. glabriusculum). Conserv Genet 3:123–129
Walsh BM, Hoot SB (2001) The phylogenetic relationships of Capsicum (Solanaceae) using DNA sequences from two non-coding regions: the chloroplast atpB-rbcL spacer region and nuclear waxy introns. Int J Plant Sci 162:1409–1418
Yi G, Lee JM, Lee S, Choi D, Kim BD (2006) Exploitation of pepper EST–SSRs and an SSR-based linkage map. Theor Appl Genet 114:113–130
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. The Plant Genome 1:5–20
Zwendie Y, Tong N, Bosland P (2004) Establishing a core collection of Capsicum using a cluster analysis with enlightened selection of accessions. Genet Res Crop Evol 51:147–151
Acknowledgments
The authors thank Ghislaine Nemouchi and Bruno Savio for technical help, Charles Poncet as coordinator of the Gentyane PlateForme of INRA-Clermont Ferrand which hosted the genotyping of the accessions, Christopher Sauvage, Nicolas Ranc (INRA-GAFL Avignon) and Marie-Hélène Muller (INRA Montpellier) for discussions and critical review of the manuscript. This work was supported by the SPICY European project (“Smart tools for Prediction and Improvement of Crop Yield”, KBBE-2008-211347).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Nicolaï, M., Cantet, M., Lefebvre, V. et al. Genotyping a large collection of pepper (Capsicum spp.) with SSR loci brings new evidence for the wild origin of cultivated C. annuum and the structuring of genetic diversity by human selection of cultivar types. Genet Resour Crop Evol 60, 2375–2390 (2013). https://doi.org/10.1007/s10722-013-0006-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-013-0006-0