
Abstract
The capybara, Hydrochoerus hydrochaeris, is a semiaquatic herbivorous rodent widely distributed in South American wetlands. Some authors have proposed the existence of two species, several subspecies, and different Evolutionarily Significant Units (ESUs) in the Hydrochoerus genus. However, the available genetic studies of capybaras are limited in their geographical scope. Thus, we examined genetic variation in 387 sequences of the mitochondrial control region (124 generated by the authors and 263 obtained from GenBank) and performed phylogeographic analyses to determine patterns of geographic variation in capybaras sampled in seven South American countries, representing a large portion of the distribution range of the species. Based on these analyses, four ESUs were identified. In addition, within one of these units, two distant geographic populations were identified in the Trans-Andean Colombia (putative H. isthmius population) and the cis-Andean area of the Napo River in Peru and Ecuador. This result and the genetic distances between these ESUs agree with the idea of a unique capybara species. Mismatch distributions and Bayesian skyline plots suggested that capybaras have undergone processes of population decline associated with the adverse weather conditions of the late Pleistocene, followed by population expansions associated with the climatic conditions of the Holocene Climatic Optimum. Phylogenetic trees support the previous hypothesis that the species originated in Western Amazon. However, as our results are based on a single mitochondrial fragment, these results are not conclusive. New studies, including samples from other sites and more mitochondrial and nuclear markers, may elucidate the evolutionarily history of capybaras.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The capybara, Hydrochoerus hydrochaeris (Linnaeus, 1766; type locality: Surinam, but restricted to San Francisco River, Sergipe state in Brazil by Feijó and Langguth 2013), is a semiaquatic herbivorous rodent widely distributed throughout most of the South American wetlands. Its geographical distribution encompasses from Panama to south of Buenos Aires Province in Argentina. The taxonomic history of capybaras is complex and some authors differentiated a second species from H. hydrochaeris. This second species, H. isthmius, occurring in eastern Panama, northwestern Venezuela, and northern and western Colombia, was first described by Goldman (1912), with a type locality in Marragantí, near the head of tide-water on the Tuira River at the Darien region, near the frontier between Panama and northern Colombia. Based on morphology (Mones 1984; Mones and Ojasti 1986) and genetics (Moreira et al. 2012), H. isthmius was recognized as a distinct species in Mammal Species of the World (Wilson and Reeder 2005). However, isozyme (Peceño 1983) and mitochondrial DNA analyses (Ruiz-García et al. 2016a, b) revealed small genetic differences between H. isthmius and H. hydrochaeris, suggesting sub-specific differences and agreeing well with Heinemann’s (1975) subspecies proposal (H. h. isthmius). Also in agreement with this last author, Ruiz-García et al. (2016b) showed that putative populations of Colombian H. isthmius were genetically more related to some Amazonian H. hydrochaeris populations than these last to another Amazonian H. hydrochaeris populations. Thus, these molecular results agree better with H. h. isthmius as a subspecies than as a differentiated species from H. hydrochaeris.
The information about the geographical distribution of H. isthmius and H. hydrochaeris and current population status varies considerably between countries and regions. Thus, the Trans-Andean species H. isthmius is reported to be common in Panama but rare in Colombia and Venezuela (Moreira et al. 2012; Delgado and Emmons 2016) and is catalogued as Data Deficient by the IUCN (Delgado and Emmons 2016), whereas H. hydrochaeris is reported as common through most of its range and is listed as Least Concerned in Brazil (ICMBio/MMA 2018), Paraguay (Saldívar et al. 2017), and Argentina (Bolkovic et al. 2019). In Uruguay, capybaras are considered a priority species (González et al. 2013).
Despite being widely distributed, the species faces many different conservation problems. In Venezuela and Colombia, the species’ current distribution is highly fragmented and has experienced population declines in many regions, even going extinct in some (Ojasti 1973; Maldonado-Chaparro et al. 2011; Herrera and Barreto 2012). In Brazil, the species is considered a local pest because of the damage it causes to crops (Paschoaletto et al. 2003) and is almost extinct in the Caatinga biome, possibly due to high hunting pressure and habitat alteration (Moreira 2004). In addition, the scarcity and even disappearance of the species in northeastern Brazil have been noted in recent decades (Paiva 1973). In Argentina, capybaras are usually abundant in protected sites, but outside of them, densities drop dramatically. In places where wetlands were transformed into cattle pastures, populations decreased dramatically or directly disappeared (Bolkovic et al. 2019).
Sequences from the first hypervariable region (HVRI) of the rapidly evolving noncoding control region (CR) have been extensively used in the last decade to analyze the genetic diversity, population structure, and historical population dynamics of the species at different geographical scales. The first study was carried out by Campos-Krauer and Wisely (2010) who performed a phylogeographic analysis of the species using non-invasive samples collected in 13 populations located in the Gran Chaco ecosystem of Paraguay. They obtained relatively low values of haplotype diversity and genetic signals of recent population range expansions caused by deforestation and cattle ranching. Borges-Landáez et al. (2012) assessed the genetic variability and population structure of five managed capybara populations from the seasonally flooded savanna of Venezuelan Llanos and found moderate to high levels of genetic diversity, a significant genetic structuring and a correlation between genetic and geographic distance, suggesting restricted gene flow with isolation by distance. Byrne et al. (2015) analyzed CR sequences from Argentina together with those available from Paraguay and Venezuela in order to test the role of river drainage in shaping the genetic structure of the species at the river basin and regional scales. At the regional scale, their results showed that populations from Venezuela, located in the Orinoco Basin, were genetically different from those in Paraguay and Argentina, which belong to the Río de la Plata basin. At the basin scale, only populations from Paraguay showed significant differences between basins and a significant correlation between the genetic distance and the geographical distance between populations measured through the rivers, while for Argentina and Venezuela, results were not significant. These results would suggest that in Paraguay, the current genetic structure of capybaras is associated with the lack of dispersion corridors through permanent rivers whereas, limited structuring in Argentina and Venezuela is likely to be the result of periodic flooding facilitating dispersion. Ruíz-García et al. (2016b) sequenced the CR and Cytochrome b in 78 wild capybaras sampled in Colombia, Peru, Ecuador, and Brazil, as well as 25 wild capybaras at ten mitochondrial genes. They found 43 haplotypes, high levels of genetic diversity, and four well-defined populations in delimited geographical areas. However, their results do not support the view that two different species of capybara are present, as it was aforementioned unless chromosomal speciation can be demonstrated between these two groups. Finally, Byrne et al. (2019) analyzed the genetic diversity, population genetic structure, and historical population dynamics of the species in the Chaco-Pampean region. Their results showed the existence of four haplogroups in the study area, low genetic diversity in three of them, evidence that support the hypothesis that the Paraná and Paraguay rivers would be acting as a migration corridor for the species, and a historical population dynamics in which population expansions and secondary contact between genetic groups would be related to past climatic events.
Mitochondrial DNA analyses should be instrumental in helping to determine conservation strategies based on the systematic classification of differentiated populations below the species level. The uncertainty about these conservation units can lead to confusion in the establishment of management plans and errors in setting priorities (O’Brien 1994; DeSalle and Amato 2004). In this sense, the present study aims to combine the capybara mitochondrial DNA control region sequences obtained by the authors in previous studies at a regional scale (n = 124) with those available to date in the GenBank database (n = 264) to analyze the phylogeographic patterns of the species, the existence of geographical limits among the populations, and the taxonomic status of the different genetic units in a large portion of its distribution area in South America.
Materials and methods
Sampling
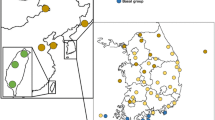
A total of 124 tissue, hair, or fecal samples were collected from different sampling sites in Colombia, Ecuador, Peru, Brazil, and Argentina (Table 1, Fig. 1). Tissue samples were obtained from animals found dead or hunted by indigene near their respective communities for food. Hair samples were obtained from live animals maintained in these communities as pets. Fecal samples were collected in the field from different mounds separated from more than 50 m, in order to decrease the probability of re-sampling the same individual. Samples were preserved in a 20% dimethyl sulfoxide solution supersaturated with NaCl or in ethanol 96% during fieldwork and stored at -20 °C until DNA extraction was performed.

Location of capybara sampling sites in (A): Venezuela, Colombia, Ecuador, Peru, and Brazil and (B): Paraguay and Argentina. Sampling site numbers correspond to those described in Table 1. Genetic groups (see results): Red-Trans-Andean (includes sampling sites in the Napo River, cis-Andean area; see discussion section for details), Yellow-Llanos, Green-Western Amazon, and Blue-Río de la Plata
Mitochondrial DNA extraction, PCR amplification and sequencing
Different DNA extraction protocols were used depending on the sample source. Tissues samples were incubated overnight at 37 °C in an extraction buffer containing 10 μl of proteinase K, 10 mg/ml; 5 μl of RNase, 20 mg/ml, and 10% SDS. After incubation, DNA was isolated by phenol–chloroform extraction and alcohol precipitation. DNA from hair follicles was extracted with 10% Chelex resin (Walsh et al. 1991). For fecal samples, we followed the protocol described in Reed et al. (1997) with one modification: in the last step of the Wizard SV Gel and PCR Clean-Up System kit (Promega) protocol, centrifugation time was changed from 1 to 5 min in order to increase the amount of purified DNA. To avoid contamination among samples, DNA was extracted in a room where no PCR products were stored.
Amplification protocols used also depended on the sample source and the quality of DNA obtained, more degraded in fecal samples. Primers, PCR mix concentrations, and PCR profiles used are described in Byrne et al. (2019) and Ruiz-García et al. (2016a, b). All amplifications including positive and negative controls were checked in 1–2% agarose gels. The amplified samples were purified using membrane-binding spin columns (Qiagen) or the EXO/SAP method (Werle et al. 1994). The double-stranded DNA was directly sequenced in a 377A (ABI) automated DNA sequencer using the same oligonucleotide primers used in PCR amplification. Some samples were repeated to ensure sequence accuracy. NUMTs were not expected because only a fragment of the mitochondrial control region was sequenced.
Control region sequences from capybaras inhabiting 13 populations located in the Gran Chaco ecosystem of Paraguay and five populations from the Venezuelan Llanos were obtained from GenBank (Fig. 1 and Table 1). With the inclusion of these sequences, the sampled area covers a large portion of the species' distribution, even though populations in most of Brazil and northeastern South America await study. Sequences from Paraguay had a length of 386 bp and were obtained from 110 individuals (Campos-Krauer and Wisely 2010; GenBank Access numbers GU456363-376), while those from Venezuela had a length of 545 bp and were obtained from 153 individuals (Borges-Landáez et al. 2012; GenBank Access Numbers EU149767- EU149776). All these sequences were fitted at a length of 292 bp due as this was the size of the HVRI fragment amplified in this study.
Data analyses
All sequences were aligned and analyzed for polymorphic sites (S) using the Clustal W system (Thompson et al. 1994) implemented in MEGA X software (Kumar et al. 2018). Parameters for the alignment were set in 15.00 for gap opening penalty and 6.66 for gap extension penalty both for pairwise and multiple alignments. The number of haplotypes (H), haplotype frequencies and haplotype (Hd), and nucleotide (π) diversities (Tajima 1983; Nei 1987) were computed using Arlequin 3.5.2 (Excoffier and Lischer 2010).
Two phylogenetic trees were obtained. The first one was a Maximum Likelihood (ML) tree constructed using RAxML v8.2.10 software (Stamatakis 2014). Best-fit models were selected using Bayesian information criteria under a ‘greedy’ search scheme using a subset of models specific to RAxML. The GTR + G model (General Time Reversible model + gamma-distributed rate variation among sites, Lanave et al. 1984) was used to search for the ML tree. We estimated support for nodes using the rapid-bootstrapping algorithm (_f a–x option) for 1,000 non-parametric bootstrap replicates (Stamatakis et al. 2008). In this first tree, no outgroup was used, in order to accommodate debated issues about molecular dating of recent phylogenetic splits (Ho et al. 2008). The second was a Bayesian tree completed with the BEAST v1.8.1 program (Drummond et al. 2012). Four independent iterations were run with six Markov Chain Monte Carlo (MCMC) chains sampled every 1,000 generations for 30 million generations after a burn-in period of three million generations. We checked for convergence using Tracer v1.6 (Rambaut et al. 2013). We plotted the likelihood versus generation and estimated the effective sample size (ESS > 200) of all parameters across the four independent analyses. The results from different runs were combined using LogCombiner v1.8.0 software (Rambaut and Drummond 2013a) and TreeAnnotator v1.8.0 software (Rambaut and Drummond 2013b). A Yule speciation model and a relaxed molecular clock with an uncorrelated log-normal rate of distribution (Drummond et al. 2006) were used. Posterior probability (PP) values provide an assessment of the degree of support of each node on the tree. The outgroup employed for rooting the Bayesian tree was the paca, Cunniculus paca.
The median-joining network method (Bandelt et al. 1999) implemented in PopART 1.7 (http://popart.otago.ac.nz) was applied to our dataset to estimate the phylogeographic structure of haplotypes. This method, using a parsimony criterion, combines the minimum-spanning trees (MSTs) with a single network, allowing more detailed population information than do strictly bifurcating trees (Posada and Crandall 1998).
After that, an Analysis of Molecular Variance (AMOVA; Excoffier et al. 1992) was performed on the four clusters identified (see Results) using Arlequin 3.5.2. The significance of the observed Ф-statistics was tested using the null distribution generated from 10,000 non-parametric random permutations of the data matrix variables. Differences in allele frequencies (FST-like) were used to compute the distance matrix for ФST. Population pairwise ФST values were also calculated using Arlequin 3.5.2 software. The Kimura 2P genetic distances (K2P) (Kimura 1980) between populations were computed in MEGA X software.
We used several methods to determine possible demographic changes across the natural history of the capybara. First, Tajima’s D (Tajima 1989) and Fu’s FS (Fu 1997) neutrality tests were carried out using Arlequin 3.5.2 software. Then, mismatch distribution analyses were performed using the same software. This method, based on an assumed stepwise growth model (Rogers and Harpending 1992), was used to evaluate: (1) whether there was a signature of population expansion, and (2) the timing of demographic expansion measured in units of mutational time. Approximate confidence intervals for growth parameters were obtained by a parametric bootstrap approach (1,000 replicates). The validity of the estimated stepwise expansion model was tested using the same parametric bootstrap approach. A goodness-of-fit test between the observed and simulated distributions of pairwise differences was performed. To quantify the smoothness of the observed haplotype frequency distribution, Harpending’s raggedness index (Harpending 1994) was applied. Statistical significance of the index was tested by a coalescent simulation (10,000 permutations) of the randomly expected value, as implemented in DnaSPv.5 (Librado and Rozas 2009). Moreover, the tau value (τ), obtained in the mismatch distribution analysis provides a rough estimate of the time when the sudden population expansion started (Rogers and Harpending 1992; Rogers 1995). The substitution rate suggested by Ruíz-García et al. (2016b) for the capybara mitochondrial control region (2.57 × 10 − 1substitutions/site/My) was used to convert mutational time (τ) into real-time according to the equation: τ = 2μt, where t is the time in years and μ is the substitution rate per generation. The generational time used was three years (Colin 1991; Nowak 1991). Finally, Bayesian skyline plots (BSP) were obtained through the BEAST 1.8.1 and Tracer 1.6 Software. The Coalescent-Bayesian skyline option in the tree priors was selected with five steps and a piecewise-constant skyline model with 40,000,000 generations (the first 4,000,000 discarded as burn-in). In the Tracer v1.6 software, the marginal densities of temporal splits were analyzed and the Bayesian Skyline reconstruction option was selected for the trees log file. A stepwise (constant) Bayesian skyline variant was selected with the maximum time as the upper 95% High Posterior Density (HPD) and the trace of the root height as the treeModel.rootHeight.
Results
The 387 individuals analyzed presented 39 different haplotypes determined by 30 polymorphic sites, of which three were singletons. Two of them were found on the sequences obtained from the 124 individuals sampled and the other in the GenBank dataset. Haplotypes 1, 19, 22, and 12 were the most frequent, being found in 74 (19.12%), 58 (14.99%), 47 (12.14%), and 41 (10.59%) individuals, respectively (Appendix I). None of the haplotypes were present in all the sampling sites. However, haplotype 1 was present in most of the sampling sites in Argentina and Paraguay; haplotypes 16, 18, and 19 were shared between Venezuela and Colombia; and haplotype 26 was shared between Ecuador and Peru. The best substitution evolutionary model was HKY + G, both for the Bayesian and Akaike models (BIC = 2038.29, AICc = 1466.15).
The ML tree showed four genetic clusters (Fig. 2). However, bootstrap values were medium or low for three of these capybara groups. The first cluster that diverged corresponds to the Western Amazon group. This group is the one with the greatest statistical support (84%). It includes eight haplotypes (n = 21 individuals); six from the Peruvian Amazon (H32-37, n = 15 individuals), and two from the Venezuelan Llanos (H38-39, n = 6 individuals) that might have an Amazonian origin. The cluster showed a high value of haplotype diversity (H = 0.876) and a relatively high value of nucleotide diversity (π = 0.011). The second cluster to diverge corresponds geographically to the Río de la Plata basin (Paraguay and Argentina) plus the central Brazilian Amazon (Negro River). This cluster included 15 haplotypes (n = 162 individuals): one shared between Paraguay and Argentina (H01, n = 74 individuals), five from Argentina (H02-06, n = 21 individuals), eight from Paraguay (H07-14, n = 66 individuals), and one from Brazil (H15, n = 1 individual). This cluster also showed a high value of haplotype diversity (H = 0.717), but lower than the previous clade, and a relatively high value of nucleotide diversity (π = 0.011). Some Paraguayan haplotypes (H09-14) appear to be the most ancestral of the cluster. Interestingly, the individual sampled in the Negro River, in the center of the Brazilian Amazonas, was associated with this cluster and not to the Western Amazon one. The last two clusters to diverge are the Trans-Andean and the Llanos. The Trans-Andean cluster included six haplotypes (n = 27 individuals): five from the Trans-Andean area of Colombia (H27-31, n = 23 individuals), and one shared between Ecuador and Peru (Cis-Andean area-Napo River) (H26, n = 4 individuals). The cluster showed a high value of haplotype diversity (H = 0.747), but a relatively low nucleotide diversity (π = 0.006). Finally, the Llanos cluster included ten haplotypes (n = 177 individuals): four from Venezuela (H21-24, n = 72 individuals), three from Colombia (H17, 20 and 25, n = 21 individuals), and the other three shared between Venezuela and Colombia (H16, 18–19, n = 84 individuals). Like the other three clusters, it presented a high value of haplotype diversity (H = 0.791) but a relatively low value of nucleotide diversity (π = 0.006) as the Trans-Andean cluster.

Maximum Likelihood (ML) tree for the 39 mitochondrial DNA control region (D-loop) haplotypes found. Nodes are labeled with bootstrap percentages. Haplotype numbers correspond to those described in Appendix I. * Calculation of the genetic diversity indexes of the Trans-Andean cluster does not include Haplotype 26. See the discussion section for details
In turn, the Bayesian tree showed two main clusters (Fig. 3), one composed of the Trans-Andean and Llanos haplotypes and the other by the Western Amazon and Río de la Plata haplotypes. Some haplotypes from Paraguay (H12-14) seem to diverge first, followed by all other haplotypes from the Río de la Plata basin and those from the Amazonian cluster. The cluster including the individuals from the Colombian and Venezuelan Llanos diverges later, followed by the Trans-Andean cluster.

A Bayesian tree for the 39 mitochondrial DNA control region (D-loop) haplotypes. Nodes are labeled with posterior probability values. Haplotype numbers correspond to those described in Appendix I. Genetic groups: Red-Trans-Andean, Yellow-Llanos, Green-Western Amazon, and Blue-Río de la Plata
The Median Joining Network (Fig. 4) showed the presence of four consecutively connected sub-networks separated by 1 to 5 mutational steps. Taking into account the position of the outgroup (d-loop sequence from the paca, Cunniculus paca) in the network, the first cluster to diverge correspond to individuals from the Colombian and Venezuelan Llanos, followed by the Western Amazon and Trans-Andean ones. The last cluster to diverge is composed of individuals from the Río de la Plata group. The star-like topology found in this cluster suggested that H01, the most frequent haplotype in the study area, represented an ancestral form from which the other haplotypes in the cluster derived. A similar pattern is observed in the other clusters, where H18, H31, and H32 seem to represent the ancestral variants.

Genealogical relationships of the 39 capybara haplotypes analyzed. Circle areas are proportional to haplotype frequencies. Each small bar on the branches represents one mutational step. Haplotype names correspond to those described in Appendix I. *The Trans-Andean cluster includes one haplotype (H26) from the Napo River of Ecuador and Peru (Cis-Andean cluster). See the discussion section for details
The results of AMOVA showed significant differences between clusters (ФST = 0.723; P = 0.0001; Table 2). The greatest source of variation (72.26%) was found between clusters. Pairwise comparisons of ФST values also showed significant differences between the four clusters (Table 2). Kimura 2P genetic distances between clusters were equal to or lower than 0.042 (Table 2).
Results of the neutrality test showed negative and non-significant (P > 0.30) Fs values for all clusters, positive and non-significant (P > 0.58) D values for the Llanos, Trans-Andean, and Western Amazon clusters, and a negative and non-significant (P = 0.48) D value for Rio de la Plata cluster (Table 3). Consequently, there is no evidence to support a demographic expansion for any cluster. However, mismatch distribution analyses showed a unimodal curve that did not differ significantly from the simulated pattern based on a recent expansion model for Llanos, Trans-Andean, and Western Amazon clusters (P > 0.11, Fig. 5a-c). Raggedness values also provided statistical support for these results (P > 0.21). The time elapsed since expansion occurred was estimated at 14,800 Years Before Present (YBP) for Llanos, 14,200 YBP for Trans-Andean, and 27,800 YBP for the Western Amazon cluster. On the other hand, the Rio de la Plata cluster showed a multimodal curve (Fig. 5d). The Bayesian Skyline Plots (Fig. 5e-g) showed that Llanos, Western Amazon, and Río de la Plata suffered a decline in their female effective population sizes at about 60,000, 300,000, and 50,000 YBP, respectively, followed by an increase in the last 10,000 to 4,000 years. For the Trans-Andean cluster, results did not show a good resolution of possible demographic changes.

Demographic history of the four capybara genetic clusters identified. Mismatch distributions (left panel): Bars are observed distributions; dotted lines are expected distribution under a sudden expansion model. Bayesian skyline plots (right panel): The black line represents the estimated median and the grey zone the 95% highest posterior density (HPD) intervals. References: Llanos (a, e), Western Amazon (b, f), Trans-Andean (c), Río de la Plata (d, g)
Discussion
Determining how species are divided into genetically distinguishable units is fundamental for conservation planning. As evolutionary processes act at the intraspecific level, genetic differences and locally adaptive characters will accumulate in these units over time. This reservoir of genetic and phenotypic diversity increases the species’ ability to persist through environmental changes (Wang 2002). Thus, one of the main goals in conservation is to preserve the evolutionary potential of species by maintaining the diversity found in genetic units. There are two levels of genetic differentiation within a species, called Evolutionarily Significant Units (ESUs) and Management Units (MUs) (Ryder 1986; Moritz 1994a, b; Vogler and Desalle 1994; Funk et al. 2012). An ESU should be reciprocally monophyletic for mitochondrial haplotypes and show significant differences at nuclear loci, while a MU is defined as any population that exchanges so few migrants with other populations as to be genetically distinct from them (Avise 2004). In practice, MUs are identified by significant differences in allele frequencies at neutral marker loci. The identification of ESUs and MUs is primarily relevant to long-term management issues, that is, defining conservation priorities and setting strategies (Moritz 1994a). In this sense, the results obtained by the Median Joining Network and the AMOVA were coincident in the existence of four different genetic groups in our study area (i.e., Llanos, Trans-Andean, Western Amazon, and Río de la Plata basin; Fig. 1), although these groups are separated by a small number of substitutions in the network. The first group includes individuals from the Eastern Colombian Llanos, the northern Colombian Amazon (Guainia Department that borders with the Eastern Colombian Llanos), and Venezuela. The second is composed of individuals from northern and western Colombia (Trans-Andean area), but also by a small population in the Ecuadorian and Peruvian Amazon area of the Napo River (Cis-Andean area). That is to say, this group includes two populations geographically distant from each other and separated by the Andes Mountains. Taking into account that both populations are in very different geographical and ecological areas, they should be separated into two different ESUs as previously suggested by Ruiz-García et al. (2016b), or in two different MUs, or geographical populations, within the ESU. Carrying out an exhaustive sampling along the Napo River would make it possible to establish the contact zone and the existence of a hybrid zone between this alleged ESU and that of the Western Amazon, which would confirm the existence of a single capybara species, such as suggested by molecular data (see below). The next group is limited to the Peruvian Amazon, while the last one includes individuals from Paraguay and Argentina and one specimen from the central Brazilian Amazon (Negro River). According to the results of the median-joining network, these groups could be considered as different ESUs, as they are reciprocal monophyletic for the mitochondrial DNA control region haplotypes analyzed. In this analysis, the Trans-Andean haplogroup (“a priori” H. isthmus) was placed intermixed with the cis-Andean haplogroups (“a priori” H. hydrochaeris), which agrees quite well with a unique species of capybara. However, in the ML and Bayesian trees, the Rio de la Plata haplotypes were clearly not monophyletic because two clusters were detected (which were not resolved in the ML tree). Additionally, the node support values were medium or low for three of these capybara haplogroups. Therefore, in the strict sense of the word, only one ESU was found, corresponding to the Western Amazon haplogroup (node support values; ML = 84; Bayesian tree = 0.97). Nevertheless, although the Trans-Andean (including the specimens from the cis-Andean area of the Napo River in Ecuador and Peru), Llanos, and Rio de la Plata haplogroups did not have elevated bootstraps or posterior probabilities, they were located in well-defined geographical areas, which have a biological conservation meaning. Probably, with the analysis of more control region sequences or other mitochondrial/ nuclear genes, the statistical significance of these haplogroups will increase, and therefore they could be considered as “true” ESUs. Our discrete bootstrap or posterior probability values were the consequence of the analysis of only one gene. Moreover, seventeen of the 39 haplotypes found in our study area are shared between sampling sites, but always between those belonging to the same putative ESU (Appendix I). These results coincide with that proposed by Byrne et al. (2015), who suggested that the capybara population from Venezuela (Orinoco River basin) should be preliminarily considered a different ESU from the populations of the Río de la Plata basin, and are mostly coincident with the four ESUs proposed by Ruiz-García et al. (2016b). There are at least two other requisites for the definition of ESUs, i.e., to show significant differences of allele frequencies at nuclear loci, and phenotypic traits, mainly variation in skull metrics, but also other characteristics such as body weight, life history, and behavior (Moritz 1994a; Frankham et al. 2010). Until now, there are only two available studies in which nuclear markers were used in capybaras (Herrera et al. 2004; Maldonado-Chaparro et al. 2011), but both are very limited in the geographic range analyzed to define genetic units. Regarding phenotypic traits, Mones and Ojasti (1986) and Moreira et al. (2012) reported the existence of a latitudinal cline in the species, with an increase in body size and mass as the latitude increases. However, the first two authors warned that subspecies recognition could only be based on extreme populations and arbitrary limits.
Regarding the taxonomic status of the capybara genetic units, Mones and Ojasti (1986) recognized H. isthmius as a distinct species from H. hydrochaeris based on anatomical differences. However, Ruiz-García et al. (2016b) found genetic distance values lower than 3% at the mitochondrial control region between the Trans-Andean (representatives of the putative H. isthmus species) and the Llanos/Western Amazon populations. The authors concluded that their results agree better with a unique species of capybara because their genetic distance values are typical of the status of different populations or even subspecies but not of different species according to Avise (1994). Our results also agree with the idea of a unique capybara species, as we found genetic distance values from 2 to 4.2% between the Trans-Andean population (putative H. isthmus) and all other populations (H. hydrochaeris). Following Avise (1994), Bradley and Baker (2001), Kartavtsev (2011), and Ruiz-García et al. (2014), the average genetic distances among full species of the same genus is around 8–11%. Clearly, the genetic distances obtained were lower. On the other hand, Cabrera (1961) recognized three capybara subspecies: H. h. dabbenei from Paraguay and northeastern Argentina, H. h. uruguayensis from Uruguay and eastern Argentina, and the nominotypical form from the remainder of the range. Results obtained from the analysis of the mitochondrial DNA control region show no evidence to support the existence of two subspecies or ESUs within the Río de la Plata basin. Most of the sampling sites in Paraguay and Argentina, which cover the geographic areas mentioned by Cabrera (1961), share the most common capybara haplotype (H01, Appendix I), indicating the lack of reciprocal monophyly among populations. Moreover, Byrne et al. (2019), analyzing the population genetic structure of the species in the Chaco-Pampean region, found a widespread haplogroup that suggests the existence of historical gene flow, possibly mediated by the Paraguay and Paraná rivers, between sampling sites located at the Gran Chaco ecosystem of Paraguay and the Chaco-Pampean region in Argentina. In summary, our results support the existence of four or five groups (potential ESUs when more molecular markers are analyzed) throughout the study area, but do not agree with the existence of the two species recognized by Mones and Ojasti (1986) or all three subspecies proposed by Cabrera (1961). However, conclusions regarding the existence of one or two capybara species based on a fragment of a single mitochondrial marker should be taken with caution. More genetic studies, complemented with morphological and ecological data, and including samples from the Darien region of Panama, whole mitochondrial genomes, and nuclear genes, may contribute to resolving this question.
Until now, several studies have estimated the genetic diversity of capybara populations using the HVRI of the mitochondrial control region (Campos-Krauer and Wisely 2010; Borges-Landáez et al. 2012; Ruíz-García et al. 2016a, b; Byrne et al. 2019). Haplotype diversity reported in these studies is highly variable (H = 0.00–0.98) as well as the sample number used to calculate it in each population. In the present study, where the 387 HVRI sequences available for the capybara were analyzed, we found high values of haplotype diversity (H = 0.717–0.876) and relatively low to high values of nucleotide diversity (π = 0.006–0.011) in the four clusters identified. Taking into account the long history of commercial exploitation and poaching of capybara populations in Argentina and Paraguay it could affect the future of these populations (Bolkovic et al. 2006; Schivo et al. 2015). The effect of poaching on genetic diversity has been reported by Maldonado-Chaparro et al. (2011) for a capybara population in the Colombian Eastern Llanos. Analyzing five microsatellite markers in 31 capybaras from Hato Corozal, Colombia, the authors found that the population crossed over a very narrow recent bottleneck, possibly caused by the illegal hunt. Besides, local extinctions mainly caused by high hunting pressure and habitat alteration have been reported in Venezuela, northeastern Brazil, and Argentina (Paiva 1973; Moreira et al. 2012; Bolkovic et al. 2019). Thus, even though the species as a whole presents high levels of genetic diversity, genetic monitoring of capybara populations and the identification of genetic units is important to guarantee adequate management plans for the species.
Regarding the demographic history of the four genetic groups identified, the results of BSP analyzes suggest that three of these groups have undergone processes of population decline in the last 300,000 to 50,000 years. Quaternary climatic fluctuations have been widely recognized as one of the main historical processes influencing the historical dynamics of natural populations both in the northern and southern hemispheres (Vuilleumier 1971; Hewitt 2011). Habitat contractions caused by Pleistocene glacial cycles have been proposed as a major process in reducing population numbers and genetic diversity in different species that inhabits temperate regions of South America (Ruzzante et al. 2006). In particular, the late Pleistocene period is characterized by arid to semiarid and cold weather conditions, with a mean decrease of temperature between 5 and 7 °C in high-altitude areas (Quattrocchio et al. 2008; Peçanha et al. 2017). These conditions are very adverse for the capybara, a species for which water is an essential resource, and its distribution is associated with temperate mean annual temperatures (Byrne 2017). In this context, it is possible that the capybara population declines would be associated with the climatic characteristics of the late Pleistocene.
Loss of genetic diversity due to genetic drift in declined populations can be followed by rapid range expansions as the weather becomes more favorable. The results obtained for the mismatch distribution and BSP analyzes suggest that this could have occurred in the four genetic groups of capybaras in a period between the end of the Pleistocene and the first half of the Holocene (ca. 28,000–4,000 YBP). For example, our data suggest that Río de la Plata population expansion would have occurred during the Holocene Climatic Optimum (7,000 to 3,000 YBP, Giorgis et al. 2015), a period characterized by heavy rainfall. This coincides with that observed by Byrne et al. (2019) who related the population expansion of Argentinean populations with the formation of the Iberá Wetland and the increase in predominance of herbaceous vegetation associated with this water body (Fernández Pacella et al. 2011) that generated a habitat with optimal conditions for capybaras (Schivo et al. 2015). Similar processes could have occurred throughout the distribution of the species, where the heavy rainfalls distinctive of the Holocene Climatic Optimum and the associated changes in vegetation have played a very important role in habitat modeling (de Vivo and Carmignotto 2004). Regional changes in climate and hydrology had similar effects on other semi-aquatic species near the end of the Pleistocene (Lopes et al. 2006, 2007; Marquez et al. 2006) (Tables 4 and 5).
The geographical origin of the species has been previously analyzed by Ruiz-Garcia et al. (2016b). Their results support the Western Amazon population as the original capybara population from which the Llanos and Trans-Andean populations derived. However, this previous study did not include the samples from Venezuela, Paraguay, and Argentina included here. When all these samples were included in the analysis, the ML tree showed that the first cluster that diverged corresponds to the Western Amazon group, agreeing with what was reported by Ruiz-Garcia et al. (2016b). Three other results of our study support the Western Amazon as the geographical origin of the species. This population shows the highest genetic diversity, the most ancient bottleneck according to the BSP results, and the oldest population expansion according to the mismatch distribution analyses. However, the Bayesian tree showed a deep divergence dividing the four or five genetic clusters into two main clades that appear genetically equidistant. Within the main clade where the Western Amazon group was placed, the first to diverge were ancestors of some individuals from Paraguay, and the Median Joining Network shows that Llanos is the genetic cluster most related to the outgroup, suggesting that this is the ancestral cluster. The different results obtained regarding which population is the ancestral one are probably due to the high genetic variance due to the use of only one mitochondrial marker. In conclusion, the results obtained when all the available samples were incorporated are not conclusive about the geographic origin of the species. New studies, including samples from sites not yet sampled and a larger number of mitochondrial and nuclear genes, could help elucidate the evolutionary history of the species.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Avise JC (1994) Molecular Markers, Natural History, and Evolution. Chapman and Hall, New York
Avise JC (2004) Molecular Markers, Natural History, and Evolution, 2nd ed. Sinauer Associates Inc., Sunderland
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Bolkovic ML, Quintana R, Cirignoli S, Perovic PG, Eberhardt A, Byrne S, Bareiro R, Porini G (2019) Hydrochoerus hydrochaeris. In: SAyDS–SAREM (eds) Categorización 2019 de los mamíferos de Argentina según su riesgo de extinción. Lista Roja de los mamíferos de Argentina, Buenos Aires. http://cma.sarem.org.ar/es/especie-nativa/hydrochoerus-hydrochaeris
Bolkovic ML, Quintana RD, Ramadori D, Elisetch M, Rabinovich J (2006) Proyecto Carpincho. Propuesta para el uso sustentable del Carpincho (Hydrochoerus hydrochaeris) en la Argentina. In: Bolkovic ML, Ramadori D (eds) Manejo de Fauna Silvestre en la Argentina, Programas de uso sustentable, Dirección de Fauna Silvestre. Secretaría de Ambiente y Desarrollo Sustentable, Buenos Aires, pp 105–199
Borges-Landáez PA, Perdomo G, Herrera EA (2012) Estructura y diversidad genética en poblaciones manejadas de Chiguire en los Llanos Venezolanos. Interciencia 37:227–233
Bradley RD, Baker RJ (2001) A test of the genetic species concept: cytochrome-b sequences and mammals. J Mammal 82:960–973. https://doi.org/10.1644/1545-1542(2001)082/0960:ATOTGS/2.0.CO;2
Byrne MS (2017) Caracterización genética, filogeografía y patrones de aptitud de hábitat en el carpincho, Hydrochoerus hydrochaeris, a distintas escalas ecológicas. Tesis Doctoral, Universidad Nacional de Luján. Luján, Buenos Aires
Byrne MS, Quintana RD, Bolkovic ML, Cassini MH, Túnez JI (2015) The role of river drainages in shaping the genetic structure of capybara populations. Genetica 645–656. https://doi.org/10.1007/s10709-015-9862-1
Byrne MS, Quintana RD, Bolkovic ML, Túnez JI (2019) Population genetics of the capybara, Hydrochoerus hydrochaeris, in the Chaco-pampean region. Mammal Biol 96:14–22. https://doi.org/10.1016/j.mambio.2019.03.008
Cabrera A (1961) Catalogo de los mamíferos de América del Sur. Rev Mus Argent Cienc Nat 4:309–732
Campos-Krauer JM, Wisely SM (2010) Deforestation and cattle ranching drive rapid range expansion of capybara in the Gran Chaco ecosystem. Global Change Biol 1–13. https://doi.org/10.1111/j.1365-2486.2010.02193.x
Colin AC (1991) Reproductive biology of captive capybara. J Mammal 72:206–208
Delgado C, Emmons L (2016) Hydrochoerus isthmius. The IUCN Red List of Threatened Species 2016:e.T136277A22189896. Available from https://doi.org/10.2305/IUCN.UK.2016-2.RLTS.T136277A22189896.en. (accessed June 2021)
DeSalle R, Amato G (2004) The expansion of conservation genetics. Nat Rev Genet 5(9):702–712. https://doi.org/10.1038/nrg1425
de Vivo M, Carmignotto AP (2004) Holocene vegetation change and the mammal faunas of South America and Africa. J Biogeogr 31: 943–957. https://doi.org/10.1111/j.1365-2699.2004.01068.x
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88. https://doi.org/10.1371/journal.pbio.0040088
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. https://doi.org/10.1093/molbev/mss075
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. https://doi.org/10.1093/genetics/131.2.479
Feijó A, Langguth A (2013) Mamíferos de médio e grande porte do nordeste do Brasil: distribucao e taxonomía, com descricao de novas especies. Rev Nordest Biol 22:3–225
Fernández Pacella L, Garralla S, Anzótegui L (2011) Cambios en la vegetación durante el Holoceno en la región Norte del Iberá, Corrientes, Argentina. Rev Biol Trop 59:103–112
Frankham RJ, Ballou JD, Briscoe DA (2010) Introduction to Conservation Genetics, 2nd edn. Cambridge University Press, Cambridge
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925. https://doi.org/10.1093/genetics/147.2.915
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489-496. https://doi.org/10.1016/j.tree.2012.05.012
Giorgis MA, López ML, Rivero D, Cingolani AM (2015) Cambios climáticos en las sierras de Córdoba (Argentina) durante el Holoceno. Aportes a las reconstrucciones climáticas a través del análisis de silicofitolitos del sitio arqueológico El Alto 3. Bol Soc Argent Bot 50:361–375. https://doi.org/10.31055/1851.2372.v50.n3.12526
Goldman EA (1912) New mammals from eastern Panama. Smithsonian Misc Coll 60:1–18
González EM, Martínez-Lanfranco JA, Juri E, Rodales AL, Botto G, Soutullo A (2013) Mamíferos. In: Soutullo A, Clavijo C, Martínez-Lanfranco JA (eds) Especies prioritarias para la conservación en Uruguay. Vertebrados, moluscos continentales y plantas vasculares. snap/dinama/mvotma y dicyt/mec, Montevideo, pp 175–207
Harpending HC (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol 66:591–600
Heinemann D (1975) Superfamily: cavies. In: Grizmek B (ed) Animal Life Encyclopedia 11 (Mammals II). Van Nostrand Reinhold, New York, pp 441–453
Herrera EA, Barreto GR (2012) Capybaras as a source of protein: utilization and management in Venezuela. In: Moreira JR, Ferraz KMPMB, Herrera EA, Macdonald DW (eds) Capybara: Biology, Use and Conservation of an Exceptional Neotropical Species. Springer, New York, pp 305–319
Herrera EA, Chemello ME, Lacey EA, Salas V, Sousa BF (2004) Characterization of microsatellite markers from capybaras, Hydrochoerus hydrochaeris (Rodentia: Hydrochoeridae). Mol Ecol Notes 4:541–543. https://doi.org/10.1111/j.1471-8286.2004.00705.x
Hewitt GM (2011) Quaternary phylogeography: the roots of hybrid zones. Genetica 139(5):617–638. https://doi.org/10.1007/s10709-011-9547-3
Ho SYW, Saarma U, Barnett R, Haile J, Shapiro B (2008) The effect of inappropriate calibration: three case studies in molecular ecology. PLoS ONE 3(2):e1615. https://doi.org/10.1371/journal.pone.0001615
ICMBio/MMA (2018) Livro Vermelho da Fauna Brasileira Ameaçada de Extinção: Volume I. Ministerio do meio ambiente, Brasília
Kartavtsev Y (2011) Divergence at Cyt-b and Co-1 mtDNA genes on different taxonomic levels and genetics of speciation in animals. Mitochondrial DNA 22:55–65. https://doi.org/10.3109/19401736.2011.588215
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120. https://doi.org/10.1007/BF01731581
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lanave CG, Preparata C, Saccone C, Serio G (1984) A new method for calculating evolutionary substitution rates. J Mol Evol 20:86–93. https://doi.org/10.1007/BF02101990
Librado P, Rozas J (2009) DnaSP v.5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Lopes IF, Brito RA, Henrique-Silva FH, Del Lama NS (2006) Demographic history of wood stork (Mycteria americana) Brazilian Pantanal colonies revealed by mitochondrial DNA analysis. Genet Mol Biol 29(2):241–250. https://doi.org/10.1590/S1415-47572006000200008
Lopes IF, Miño CI, Del Lama SN (2007) Genetic diversity and evidence of recent demographic expansion in waterbird populations from the Brazilian Pantanal. Braz J Biol 67(suppl.4):849–857. https://doi.org/10.1590/S1519-69842007000500007
Maldonado-Chaparro A, Bernal-Parra LM, Forero-Acosta G, Ruiz-García M (2011) Estructura genética de un grupo de capibaras, Hydrochoerus hydrochaeris (Rodentia:Hydrocheridae) en los Llanos orientales colombianos. Rev Biol Trop 59(4):1777–1793
Marquez A, Maldonado JE, Gonzalez S, Beccaceci MD, Garcia JE, Duarte JMB (2006) Phylogeography and Pleistocene demographic history of the endangered marsh deer (Blastocerus dichotomus) from the Rio de la Plata Basin. Conserv Genet 7:563–575. https://doi.org/10.1007/s10592-005-9067-8
Mones A (1984) Estudios sobre la familia Hydrochoeridae. XIV. Revisión sistemática. Senckenb Biol 65:1–17
Mones A, Ojasti J (1986) Hydrochoerus hydrochaeris. Mammal Species 264:1–7
Moreira JR (2004) Avaliação do atual estado de uso e da conservação da capivara no Brasil. In: XLI Reunião Anual da Sociedade Brasileira de Zootecnia. Embrapa Gado de Corte, Campo Grande, pp 517–527
Moreira JR, Alvarez MR, Tarifa T, Pacheco V, Taber A, Tirira DG, Herrera EA, Ferraz KMPMB, Aldana-Domínguez J, Macdonald DW (2012) Taxonomy, natural history and distribution of the capybara. In: Moreira JR, Ferraz KMPMB, Herrera EA, Macdonald DW (eds) Capybara: Biology, Use and Conservation of an Exceptional Neotropical Species. Springer, New York, pp 3–37
Moritz C (1994a) Defining evolutionarily significant units for conservation. Trends Ecol Evol 9:373–375. https://doi.org/10.1016/0169-5347(94)90057-4
Moritz C (1994b) Applications of mitochondrial DNA analysis in conservation: a critical review. Mol Ecol 3:401–411
Nei M (1987) Molecular Evolutionary Genetics. Columbia University Press, New York
Nowak RM (1991) Walker’s Mammals of the World, 5th ed. Johns Hopkins University Press, Baltimore
O’Brien SJ (1994) A role for molecular genetics in biological conservation. Proc Natl Acad Sci USA 91:5748–5755. https://doi.org/10.1073/pnas.91.13.5748
Ojasti J (1973) Estudio biologico del chigüire o capibara. FONAIAP, Caracas
Paiva MP (1973) Distribuição e abundância de alguns mamíferos selvagens no Estado do Ceará. Ciênc Cult 25:442–450
Paschoaletto K, Ferraz M, Lechevalier M, Zarate di Couto H, Martins L (2003) Damage caused by capybaras in a corn field. Sci Agric 60:191–194. https://doi.org/10.1590/S0103-90162003000100029
Peçanha WT, Althoff SL, Galiano D, Quintela FM, Maestri R, Gonçalves GL, Freitas TRO (2017) Pleistocene climatic oscillations in Neotropical open areas: refuge isolation in the rodent Oxymycterus nasutus endemic to grasslands. PLoS One 12(11):e0187329. https://doi.org/10.1371/journal.pone.0187329
Peceño MC (1983) Estudio citogenético y genético evolutivo del chigüire género Hydrochaeris. Trabajo Especial de Grado, Universidad Simón Bolívar, Caracas
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14(9):817–818. https://doi.org/10.1093/bioinformatics/14.9.817
Quattrocchio ME, Borromei AM, Deschamps CM, Grill SC, Zavala CA (2008) Landscape evolution and climate changes in the late Pleistocene–Holocene, southern Pampa (Argentina): evidence from palynology, mammals and sedimentology. Quatern Int 181:123–138. https://doi.org/10.1016/j.quaint.2007.02.018.
Rambaut A, Drummond AJ (2013a) LogCombiner v1.8.0. http://beast.bio.ed.ac.uk/
Rambaut A, Drummond AJ (2013b) TreeAnnotator v1.8.0. http://beast.bio.ed.ac.uk/
Rambaut A, Suchard MA, Xie W, Drummond AJ (2013) Tracer v1.6 http://tree.bio.ed.ac.uk/software/tracer/
Reed JZ, Tollit DJ, Thompson PM, Amos W (1997) Molecular scatology: the use of molecular genetic analysis to assign species, sex and individual identity to seal faeces. Mol Ecol 6:225–234. https://doi.org/10.1046/j.1365-294x.1997.00175.x
Rogers AR (1995) Genetic evidence for a Pleistocene population expansion. Evolution 494:608–615. https://doi.org/10.1111/j.1558-5646.1995.tb02297.x
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569. https://doi.org/10.1111/j.1558-5646.1995.tb02297.x
Ruiz-García M, Luengas-Villamil K, Leal L, Bernal-Parra LM, Shostell JM (2016a) Phylogenetics and phylogeography of two large Neotropical rodents (Capybara, Hydrochoerus hydrochaeris, Hydrochaeridae and Paca, Cunniculus paca, Agoutidae; Rodentia) by means of mitochondrial genes: opposite patterns. In: Urbano KV (ed) Advances in Genetics Research, vol 16. Nova Science Publisher, New York, pp 151–159
Ruíz-García M, Luengas-Villamil K, Pinedo-Castro M, Leal L, Bernal-Parra LM, Shostell JM (2016b) Continuous Miocene, Pliocene and Pleistocene influences on mitochondrial diversification of the Capybara (Hydrochoerus hydrochaeris; Hydrochoeridae, Rodentia): incapacity to determine exclusive hypotheses on the origins of the Amazon and Orinoco diversity for this species. J Phylogenet Evol Biol 4(2):1–20. https://doi.org/10.4172/2329-9002.1000166
Ruiz-García M, Pinedo-Castro M, Shostell JM (2014) How many genera and species of woolly monkeys (Atelidae, Platyrrhine, Primates) are there? First molecular analysis of Lagothrix flavicauda, an endemic Peruvian primate species. Mol Phylogenet Evol 79:179–198. https://doi.org/10.1016/j.ympev.2014.05.034
Ruzzante DE, Walde SJ, Cussac VE, Dalebout ML, Seibert J, Ortubay S, Habit E (2006) Phylogeography of the Percichthyidae (Pisces) in Patagonia: roles of orogeny, glaciation, and volcanism. Mol Ecol 15:2949–2968. https://doi.org/10.1111/j.1365-294X.2006.03010.x
Ryder OA (1986) Species conservation and systematics: the dilemma of the subspecies. Trends Ecol Evol 1:9–10
Saldívar S, Rojas V, Giménez D (2017) Los Mamíferos Amenazados del Paraguay. Libro Rojo de los Mamíferos del Paraguay: especies amenazadas de extinción. Asociación Paraguaya de Mastozoología y Secretaría del Ambiente. Editorial CREATIO, Asunción
Schivo F, Kandus P, Bolkovic ML, Gail Minotti P, González Trilla G, Quintana RD (2015) A habitat suitability model for capybara (Hydrochoerus hydrochaeris) at its core area in Argentina. Trop Conserv Sci 8:150–168. https://doi.org/10.1177/194008291500800113
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57:758–771. https://doi.org/10.1080/10635150802429642
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460. https://doi.org/10.1093/genetics/105.2.437
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. https://doi.org/10.1093/nar/22.22.4673
Vogler AP, Desalle R (1994) Diagnosing units of conservation management. Conserv Biol 8:354–363. https://doi.org/10.1046/j.1523-1739.1994.08020354.x
Vuilleumier BS (1971) Pleistocene changes in the fauna and flora of South America. Science 173:771–780 https://doi.org/10.1126/science.173.3999.771
Walsh PS, Metzger DA, Higuchi R (1991) Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. BioTechniques 10:506–513
Wang JY (2002) Stock identity. In: Perrin WF, Wursig B, Thewissen JGM (eds) Encyclopedia of Marine Mammals. Academic Press, San Diego, pp 1189–1192
Werle E, Schneider C, Renner M, Völker M, Fiehn W (1994) Convenient single-step, one tube purification of PCR products for direct sequencing. Nucleic Acids Res 22:4354–4355. https://doi.org/10.1093/nar/22.20.4354
Wilson DE, Reeder DAM (2005) Mammals Species of the World: A taxonomic and geographic reference, 3rd ed. Johns Hopkins University Press, Baltimore
Acknowledgements
Thanks to Kelly Luengas-Villamil for her help in the laboratory processes for the Colombian capybara. Also thanks to some indigene communities that helped to obtain samples of capybara in Colombia (Puinave, Guahibo, Pipoko), Ecuador (Kichwa, Huaorani, Shuar and Achuar), Peru (Bora, Ocaina, Shipibo-Comibo, Capanahua, Angoteros, Orejón, Cocama), and Brazilian Amazon (Dessana). This work was supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (PIP 11420100100189 to J.I.T); Dirección de Fauna de la Secretaria de Agricultura y Desarrollo Sustentable; the Departamento de Ciencias Básicas de la Universidad Nacional de Luján; and the Consejo Nacional de Investigaciones Científicas y técnicas (PhD Grant to M.S.B).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Appendices
Appendix I Distribution of Hydrochoerus hydrochaeris control region mitochondrial DNA haplotypes by country and locality, and the total number of individuals per haplotype (Nh). Sample sizes are given at the end of the Table (Nseq). Locality names correspond to those described in Table 1.
Appendix I. Continue…
Rights and permissions
About this article

Cite this article
Byrne, M.S., Ruiz-García, M. & Túnez, J.I. Phylogeography of the capybara, Hydrochoerus hydrochaeris, in a large portion of its distribution area in South America. J Mammal Evol 29, 191–206 (2022). https://doi.org/10.1007/s10914-021-09569-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10914-021-09569-2