
Abstract
Accounting for population structure to minimize spurious associations in association analyses is of crucial importance. With sorghum genomic sequence information being available, there is a growing interest in performing such association studies for a number of important agronomic traits using a candidate gene approach. The aims of our study were to conduct a systematic survey of molecular genetic diversity and analyze the population structure in cultivated sorghum [Sorghum bicolor (L.) Moench] accessions from West Africa. Our analysis included 219 West African cultivated sorghum accessions with differing maturity intended for a marker-trait association study. A total of 27 SSRs were used, which resulted in detection of 513 alleles. Genetic diversity estimates for the accessions were found to be high. The accessions were divided into two subgroups using a model-based approach. Our findings partly agree with previous studies in that the guinea race accessions could be distinguished clearly from other accessions included in the analysis. Race and geographical origin of the accessions may be responsible for the structure we observed in our material. The extent of linkage disequilibrium for all combinations of SSRs was in agreement with expectations based on the mating system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Information on genetic diversity and population structure is a prerequisite for efficient breeding programs as it assists plant breeders in the selection of parents for crossing, providing a more rational basis for expanding the gene pool, and for identifying materials that harbor genes of value for plant improvement. Furthermore, knowledge of population structure of genetic resources is necessary for the development of strategies for appropriate conservation of genetic diversity. Therefore, in recent decades a primary activity in genetic resource management has been to characterize the structure of diversity within crop species (Brown 1989). Increasing attention has been given to dissecting and understanding diversity in relation to genes underlying important agronomic traits in a number of crops (Buckler and Thornsberry 2002). This includes several important crops such as maize (Zea mays L.), rice (Oryza sativa L.), wheat (Triticum aestivum L.), barley (Hordeum vulgare L.), and sorghum. Sorghum is an annual, predominantly autogamous cereal (Ollitrault et al. 1997) and was domesticated in northeastern Africa. Cultivated sorghum (Sorghum bicolor ssp. bicolor) has been grouped into five basic botanical races (bicolor, caudatum, durra, guinea, and kafir) and ten intermediate ones (Harlan and de Wet 1972). These races can be recognized based on their grain and spikelet/panicle morphology and can be traced back to their specific environments and the nomadic peoples that first cultivated them (Smith and Frederiksen 2000).
Sorghum is adapted to hot and dry climates as found in West and Central Africa (WCA) where it is difficult to grow most other food grains. African sorghum production is centered in the Savannah zone of WCA, where the grain of this crop is a major component of the diet for millions of people (Purseglove 1985). Apart from grains for food consumption, sorghum stover may also be used as building material or as cooking fuel and is often used to feed cattle in many West African countries (de Vries and Toenniessen 2001). Therefore, sorghum is a pillar of food security in the semi-arid zones of WCA. Worldwide it is the fifth most important cereal crop grown over an area of 43.8 million hectares (FAOSTAT 2007).
Because of its agronomic importance, there is a worldwide growing interest in characterizing the levels of genetic diversity present within sorghum using phenotypic traits, but also molecular markers (Djè et al. 2000; Grenier et al. 2000; Ghebru et al. 2002; Folkertsma et al. 2005; Deu et al. 2008). Among molecular markers most widely used in diversity studies, simple sequence repeats (SSRs) represent an ideal marker system due to their codominant inheritance, locus specificity, and multi-allelic character. Therefore, SSRs have been established as popular genetic markers for characterizing genetic variation in many plant species (Goulão et al. 2001). With the alignment of the sorghum genome sequence based on shotgun sequencing (Paterson et al. 2009), much information is available to characterize genes associated with both qualitative and quantitative variation. Association studies based on linkage disequilibrium (LD) i.e. the nonrandom associations of alleles at different loci (Flint-Garcia et al. 2003) have been performed to determine marker-trait associations in various crop plants such as oat, rice, maize, sea beet, barley (Kraakman et al. 2004), and sorghum (Casa et al. 2008). It is possible to identify genetic regions (if LD extends for the distance of several centiMorgans) or genes (if LD decays within a few thousand base pairs) associated with a particular trait of interest by genome-wide scans for genomic regions or by individual SNPs (single nucleotide polymorphisms) or SNP haplotypes within a candidate gene (Szalma et al. 2005). While performing these marker-trait association studies, ‘population structure’ may give rise to ‘spurious associations’ that is, associations between a phenotype and a marker that is not linked to any causative locus (Lander and Schork 1994). Therefore, knowledge of population structure and kinship in association studies is critical (Yu and Buckler 2006).
In WCA, photoperiod sensitivity of the local genotypes is a key adaptation trait as it assures flowering before the end of the rainy season independent of variable dates of sowing (Vaksman et al. 1996). Due to the diversity in production environments as well as large year-to-year variation in climatic conditions, formal sorghum breeding efforts in WCA that have emphasized on non-sensitive sorghum for “wide adaptation” have had limited success. Modern cultivars of sorghum with improved yield potentials may occupy specific niches in the production systems of this region, but they cannot be adopted across larger areas in WCA. The reasons for this were: firstly, the focus on early maturity in the wake of the drought periods in the 1980’s led to a removal of photoperiodic sensitivity from much of the breeding material in e.g. millet (Niangado 2001) and secondly, lack of adaptation of improved varieties to low-input conditions and climatic diversity, vulnerability to bird damage, and other biotic constraints (Omanya 2005). Hence, there is a need to incorporate variability from photoperiod sensitive sorghum to deal with these problems. Our objectives were to (1) analyze the population structure in a set of sorghum accessions from WCA differing in their photoperiod sensitivity as a pre-processing step for a candidate gene association study of genes involved in photoperiod pathway of flowering time, (2) assess the level of genetic diversity among these accessions, and (3) determine the magnitude of LD between pairs of SSR marker loci in these accessions.
Materials and methods
Plant material
Field experiments were conducted within the framework of a larger project aimed at assessing marker-trait associations for genes involved in photoperiod pathway of flowering time in sorghum. During the rainy season of 2007, a total of 219 cultivated inbred accessions of sorghum were grown at the regional research station of the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT) at Samanko, Mali. The accessions were chosen to represent a broad phenotypic diversity of photoperiodic response to flowering and were grouped into early, intermediate, and late flowering accessions based on previous analyses. It was projected that the collection should represent all sorghum material adapted to the different growing regions in WCA. This included the countries from Western West Africa (drier Savanna) like Burkina Faso, Mali, Niger, and Senegal and countries like Nigeria, Cameroon and Chad Eastern West Africa (wetter Savanna). A small proportion of accessions (n = 10) also originated from non-West African countries such as China, Ethiopia, India, Malawi, Nepal, Sudan, USA, Zambia, and Zimbabwe. The material included basic races like bicolor, caudatum, durra, and guinea. Intermediate races included in the material were durra-caudatum, kaura which is a dominating sub-race of durra-caudatum in Northern Nigeria, and breeding products from guinea × caudatum inter-racial crosses. The guinea accessions were primarily obtained from ICRISAT’s guinea core collection and included a photoperiod insensitive converted guinea accession with a short growth habit (Folkertsma et al. 2005). The inter-racial guinea × caudatum breeding lines were obtained from the Rural Economy Institute (IER), in Mali, and were comprised of sterility maintaining (B) and fertility-restoring (R) parents of the hybrid breeding program.
Genotyping
DNA extraction and polymerase chain reactions
For each accession, a leaf sample was taken for DNA extraction from the field experiment described above. The leaf material was cleaned with distilled water and dried in an oven at 40°C for 3 days. Total genomic DNA was extracted from leaf tissue using a CTAB protocol (Saghai Maroof et al. 1984) with modifications. Polymerase chain reaction amplifications of the SSRs were performed in 10 μl reaction volumes consisting of 2 μl (20–25 ng) genomic DNA, 1.5 μM of MgCl2, PCR buffer with 1 × 1.5 μM MgCl2, 0.2 μM per dNTPs, 250 μM of each primer, and 0.5 U/10 μl of HotTaq DNA polymerase (Fermentas Life Sciences). Amplifications were performed using MJ Research Thermal Cyclers (MJ Research, Watertown, MA). The cycling protocol consisted of 35 cycles with an initial denaturing step of 10 min at 95°C followed by 34 cycles at 95°C for 1 min for denaturation, followed by primer-specific annealing for 1 min and extension at 72°C for 1 min. After 35 cycles, there was a final extension step of 10 min at 72°C. Amplified PCR products were denatured and size-fractioned using capillary electrophoresis on a MegaBACE sequencer (Amersham Biosciences, Sweden). The MegaBACE Fragment Profiler v1.2 (Amersham Biosciences, Sweden) was used to size peak patterns, using internal standard ROX 400 HD for allele calling.
Simple Sequence Repeat (SSR) Markers
A total of 27 SSR primer pairs detecting single-copy loci were used to genotype the sorghum accessions (Table 1). These primer pairs have been used in earlier studies (Brown et al. 1996; Taramino et al. 1997; Bhattramakki et al. 2000; Kong et al. 2000; Schloss et al. 2002; Ramu et al. 2009), or were more recently developed to provide broader genome coverage for sorghum diversity analyses and marker-assisted selection. They were selected primarily based on the genome coverage they provide and robustness of their allele calls across laboratories (http://www.sat.cirad.fr/sat/sorghum_SSR_kitsat.kit/) and secondarily on information content (Casa et al. 2005). Out of 27 SSR markers we used, nine were genomic, 14 were EST-based and for the remaining four, their exact genomic locations were not known. For each primer pair, forward primer was labeled with fluorescent dyes 6-carboxyfluorescein (FAM), terachloro-6-carboxyfluorescein (TET) or hexachloro-6-carboxyfluorescein (HEX) and reverse primers were left unlabeled (Biomers GmbH, Germany).
Statistical analyses
The average number of alleles per locus and the number of group-specific alleles were determined for the germplasm groups and for various subsets within this collection. We calculated the allelic richness (R s ) for the races with more than ten accessions using the rarefraction method (Kalinowski 2004) by randomly sampling 10 genotypes 1,000 times and estimating the mean number of alleles per locus. The total gene diversity (H e ) was decomposed into gene diversity estimates between individual lines within each germplasm group according to Nei (1987):
where, P i is the frequency of the ith allele, and n is the number of samples. Confidence intervals for gene diversity estimates were obtained by a bootstrap procedure with resampling across markers.
The polymorphic information content (PIC) for each marker was determined using the following equation:
where P i is the frequency of the ith allele, and n is the number of alleles (Botstein et al. 1980).
A model-based approach implemented in the software package STRUCTURE (Pritchard et al. 2000) was used to determine the presence of population structure and assign sorghum accessions to subgroups. In our investigations, the set of 219 entries was analyzed by setting the number of subgroups from 1 to 20 with five runs, using the method allowing for the admixture, correlated allele frequencies and no recombination information. For each run of STRUCTURE, the burn-in time as well as the iteration number for the Markov chain Monte Carlo algorithm was set to 100,000. Evanno et al. (2005) criteria were used to decide about the best K value. Accessions with membership probabilities less than 0.80 for both subgroups were assigned to a mixed group (cf. Stich et al. 2005). Fisher’s exact test was performed in order to test disequilibrium between earliness groups of the accessions and the subgroups identified by STRUCTURE.
A permutation test using a Monte Carlo procedure was applied to test for LD between pairs of SSR loci on a genome-wide scale (Lewis and Zaykin 2002; Weir 1996). An adaptive permutation procedure was used to reduce the computational effort. If the P value was smaller than 0.3 after 2,500 permutations, another 14,500 permutations were performed. If the P value was smaller than 0.075, another 933,000 permutations were performed. LD between pairs of polymorphic loci mapped on the same chromosome (intrachromosomal comparisons) and on different chromosomes (interchromosomal comparisons) as well as r 2 (square of correlation of the allele frequencies) was evaluated for the subgroups identified by STRUCTURE. Since the power of the test for LD depends on the number of accessions analyzed per group, we used a resampling strategy to obtain comparable estimates: random samples were drawn from the group with the higher number of lines using a sample size equal to the number of lines of the smaller group. This procedure was repeated 25 times and the results were averaged (cf. Stich et al. 2005). Genetic diversity analyses was performed with software plabsoft (Maurer et al. 2004), which is implemented as an extension of the statistical software R (Ihaka and Gentleman 1996).
Results
Description of genetic diversity
Genotyping of the 219 cultivated sorghum accessions from West Africa with 27 single-copy SSR markers identified a total of 523 alleles. The number of alleles per locus ranged from 3 to 44, with an average of 19 alleles per locus (Table 2). The PIC estimates for the 27 SSR markers ranged from 0.13 to 0.94, with an average of 0.70. The highest number of alleles per locus was detected for SSR Xtxp141 (A t = 44). It was observed that SSRs with di-nucleotide repeats detected higher numbers of alleles than those with more than two repeated nucleotides. The mean number of alleles per locus for genomic SSRs (A t = 23) was higher than for EST-based SSRs (A t = 16), but was only slightly higher than that of the SSRs of unknown genomic location (A t = 24). Mean polymorphic information content for the genomic SSRs (PIC = 0.74) was higher than for both SSRs of unknown genomic location (PIC = 0.70) and EST-based (PIC = 0.67) SSRs.
It was observed that guinea race accessions had the highest number of alleles per locus (14.8) compared to the other basic races in this study. Basic race caudatum and its intermediate races with guinea and durra showed between 5.23 and 7.18 alleles per locus (Table 3). Accessions belonging to basic race kaura showed 3.55 alleles per locus. Gene diversity for race guinea was higher than other basic races (H e = 0.67). Race kaura showed lowest gene diversity among all the races (H e = 0.53). Gene diversity estimates for races caudatum and durra were found to be similar (H e = 0.56). Guinea-caudatum intermediate race accessions showed slightly higher gene diversity (H e = 0.67) than durra-caudatum race accessions (H e = 0.60), but similar to that of basic race guinea accessions. Mean allelic richness was found highest for race guinea (4.33) followed by races durra-caudatum (3.80), guinea × caudatum-bred (3.78), and caudatum (3.44).
STRUCTURE analysis
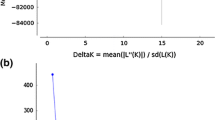
The STRUCTURE simulation demonstrated that LnP(D) value showed no clear peak for K values between 1 and 20 (data not shown). Thus, it was difficult to determine the true K (number of subgroups) using that measure. The magnitude change of LnP(D) relative to the standard deviation, called ΔK (Evanno et al. 2005), showed its highest peak at K = 2 (Fig. 1; Table 4). We also checked the structure patterns in 10 repeats of each K value from 2 to 9 (data not shown). Figure 2 provides a graphic representation of the estimated membership coefficients to the two clusters for each individual identified by a color, obtained by running STRUCTURE, setting K = 2. A clear and stable population structure was found with this approach. Subgroup one consisted of 64 and subgroup two of 109 accessions; 46 accessions had for both subgroups membership probabilities of less than 0.80, and thus, were assigned to a mixed group. After coding the sorghum accessions from each STRUCTURE subgroup by their race and maturity class (Fig. 2) it was revealed that subgroup one was dominated by the race guinea primarily belonging to early to intermediate maturity classes. The result of fisher’s exact test indicated that there were significantly (alpha = 0.06) more early genotypes in subgroup one than in subgroup two. Unlike subgroup one, subgroup two was rather mixed involving all the major accessions of sorghum belonging to intermediate to late maturity classes. After coding the accessions by their geographical origin (Fig. 3) it was found that in subgroup one, the accessions mainly originated from Burkina Faso and Mali and in subgroup two, the majority of the accessions originated from countries such as Nigeria, Niger and Mali.

Magnitude of ΔK for each K value

Summary plot of estimated population structure of 219 inbred sorghum accessions for K = 2. The probability of membership of every accession to each population is sorted by membership coefficients and identified by color. Each accession is represented by a vertical column divided into K colors. G = Guinea, C = Caudatum, GC = Guinea-caudatum, GC-bred = Guinea × Caudatum bred, CD = Caudatum × Durra, K = Kaura, C-bred = Caudatum bred; B = Bicolor; D = Durra; M = GM = Margaritiferum subgroup of basic race Guinea. Race and maturity class and the STRUCTURE subgroups are shown. The letters at the bottom of each bar denote to the maturity class of each accession where E: early, I: Intermediate, and L: late maturing

Distribution of major races of sorghum accessions among the subgroups identified by STRUCTURE. Country names are given below each bar, BF: Burkina Faso, SEN: Senegal, NIG: Nigeria CAM: Cameroon, WCA: Western Central Africa, TAN: Tanzania, and ZIM: Zimbabwe
Extent of linkage disequilibrium among SSR loci
Linkage disequilibrium was assessed for all combinations of SSR loci. An overview of the percentage of loci pairs in LD and magnitude of r 2 values with regard to the subgroups is given in Table 5. The percentage of locus pairs in significant (P < 0.05) LD was slightly higher for interchromosomal combination than intrachromosomal combinations when the complete set of 219 accessions was considered. The mean r 2 value was slightly higher for intrachromosomal combinations than interchromosomal combinations. The percentage of interchromosomal locus pairs in significant LD for the subgroups ranged from 16.6 to 49% and was higher than intrachromosomal LD values for all subgroups. The r 2 values for the subgroups ranged from 0.017 to 0.026. Subgroup two showed threefold more intrachromosomal and twofold more interchromosomal LD than did subgroup one. But subgroup one, showed based on r 2 values higher intra- as well as interchromosomal LD than subgroup two.
Discussion
Genetic diversity
The average number of alleles per locus detected in our study (A t = 19) was higher than found in previous studies by Djè et al. (1999), Ghebru et al. (2002), and Deu et al. (2008) (A t = 10.4, 11, and 13.9, respectively). Possible reasons for this were, firstly, that in our study the material was compiled in such a way that it represented diverse sorghum materials adapted to the different agro-ecological conditions in West Africa. Furthermore, our study included a higher proportion of guinea race, which showed higher genetic diversity- compared to the study of Folkertsma et al. (2005) using Southern and Eastern African guinea germplasm which has been reported to be less diverse than West African guinea (de Wet et al. 1972; Doggett 1988). Secondly, among the SSRs we used, there were more di-nucleotide microsatellites (above 50%) than the studies of Djè et al. (1999) and Deu et al. (2008). Furthermore, we used a large proportion of Xgap and Xtxp series SSRs that were isolated from genomic libraries (Brown et al. 1996; Bhattramakki et al. 2000; Kong et al. 2000). Therefore, these markers are more likely to include non-coding regions and hence they detected more alleles (A t = 23) leading to a higher average number of alleles per locus. The average gene diversity found in our study (H e = 0.70) was slightly higher than reported by Deu et al. (2008) for 484 sorghum varieties from Niger (H e = 0.60), and slightly lower than reported by Ghebru et al. (2002) for 28 Eritrean sorghum landraces (H e = 0.78), and Djè et al. (1999) for 120 Moroccan sorghum landraces (H e = 0.84). One reason for the higher H e values found in our study compared to Deu et al. (2008) may be due to the fact that the accessions of our study covered entire WCA, whereas sorghum collection of Deu et al. (2008) was limited to Niger. Furthermore, Deu et al. (2008) used two more Xcup SSRs, which are assumed to be located within or near genes, and indeed revealed fewer alleles (4.63 alleles per locus). In addition, the collection of Deu et al. (2008) had a higher percentage (24%) of accessions of race durra (which showed lowest gene diversity (H e = 0.39)), compared to our study (1.4%), and therefore this might have resulted in lower diversity values. Another possible reason for higher gene diversity found in the study of Ghebru et al. (2002) in comparison with our study may be that Eritrean sorghums (mainly caudatum and durra) have been investigated which seem to be exceptionally polymorphic because (1) most of the sorghum is maintained in the field as farmers’ varieties, (2) they show high levels of outcrossing and heterozygosity, (3) and little selection is practiced by the farmers leading to lower level of allelic fixation. Therefore, Eritrean sorghum exhibits more size variation within 28 landraces than observed in hundreds of sorghum landraces of the world collection (Ghebru et al. 2002). Compared to the study of Deu et al. (2008) allelic richness for the race guinea in our study was higher (R s = 4.33) but lower for the race caudatum. Based on the gene diversity estimates in our study, race guinea was the most diverse and race kaura was the least diverse race than other basic races.
Although we have found a high degree of genetic diversity present among accessions of sorghum in our study that may be similar to the above-mentioned studies, no direct comparison between the studies can be made, because estimates of gene diversity vary depending on the sampling schemes (single plant or bulked DNA), number and type of SSRs used, numbers of repeats of those SSRs, and their location in the genome i.e., coding or non-coding DNA regions (Deu et al. 2008). We detected 3–44 alleles per SSR locus in our analyses. These differences between numbers of alleles per locus detected among different races by SSRs can also be due to the mechanisms that generate variability for these different categories of DNA markers, e.g. point mutation and insertion-deletion for all DNA markers, plus polymerase slippage and/or unequal crossing-over for microsatellite markers (Grenier et al. 2000).
Population structure
Using the computer software STRUCTURE to infer population structure and employing Evanno’s method to estimate the number of subgroups, we found that our sorghum material was divided into two subgroups (Fig. 2). The STRUCTURE approach we used is different from a distance-based phylogenetic algorithm, such as neighbor joining used in previous studies (e.g. Deu et al. 2008), as STRUCTURE uses a model-based Bayesian clustering approach to assign individuals to the subgroups. The clusters identified using distance-based algorithms may be heavily dependent on both the distance measure and graphical representation chosen. Also, it is difficult to incorporate additional information such as the geographic sampling locations of individuals (Pritchard et al. 2000).
Between the two subgroups detected by STRUCTURE in our study, subgroup one mostly included accessions of race guinea (83%) while subgroup two showed a much lower proportion of guinea race accessions (28%) and additionally included other major races of our study such as caudatum and caudatum intermediate accessions (durra caudatum and guinea caudatum) and accessions of race durra and kaura. Similar observations were made in previous studies (Djè et al. 2000; Folkertsma et al. 2005) which reported that, in most cases, guinea-race sorghum accessions can clearly be distinguished from all other races. Similar to subgroup two, lack of clear distinction among races of sorghum was observed by Morden et al. (1989) and Djè et al. (2000) where 9–14% of the total variation detected was between sorghum races, which made it apparent that the races in subgroup two of sorghum were not substantially differentiated genetically. Another important observation was that Fisher’s exact test revealed that there are significantly (alpha = 0.06) more early maturing accessions in subgroup one than subgroup two. This can partly explain the structuring we found among sorghum accessions of our study. Coding sorghum accession by their country of origin (Fig. 3) showed clearly that subgroup one contained mainly guinea accession from western WCA such as Mali and Burkina Faso. The majority of these accessions showed early and intermediate maturity. In the case of subgroup two, countries as well as race-wise patterns were apparent. The accessions from countries like Nigeria and Niger were predominant followed by accessions from Mali. The accessions from Nigeria distinctively included mostly late maturing intermediate races like durra-caudatum and guinea-caudatum and sub-race kaura. The accessions originating from Niger included all caudatum accessions which were all early maturing and the Malian accessions included late maturing guinea accessions. Therefore, we can conclude that for the subgroups in our study race as well as geographic origin seems to be important for the observed structuring pattern of the accessions.
Linkage disequilibrium
In our study the percentage of locus pairs in significant LD (P < 0.05) was higher (Table 5) than reported by Hamblin et al. (2004) using RFLP markers with 8.7% of locus pairs in significant LD for 27 sorghum accessions. The reasons for this difference may be, firstly, that RFLP markers have a lower power to detect LD because they detect lesser alleles than SSR markers, therefore, much power is lost to detect LD (see Sham et al. 2000). Secondly, we surveyed a much larger number of accessions (217) than Hamblin et al. (2004). Furthermore, the accessions of the study of Hamblin et al. (2004) involved some wild sorghum accessions, which are generally higher outbreeding (Doggett 1988) and could have caused a downward bias of LD estimates.
The extent to which linked sites will have a correlated evolutionary history is a function of effective population size and recombination rate, both of which are affected by the mating system (Nordborg 2000). Sorghum is a predominantly self-fertilizing species (estimates of out-crossing range from 2 to 73% depending on panicle type and environment (Rooney and Smith 2000; Barnaud et al. 2008), and is therefore expected to show higher levels of LD than out-crossing species like maize (Nordborg 2000). Consistent with the predicted effects of self-pollination and reduced effective population size, sorghum showed a greater extent of LD than maize. However, the mating system alone cannot be a simple predictor of high levels of LD—barley is highly self-pollinating but has similar diversity and LD as maize (Morrell et al. 2003). Non-equilibrium processes like population structure, history, and selection may explain this phenomenon.
We found in our study that subgroup two exhibited a higher level of intrachromosomal as well as interchromosomal LD than subgroup one based on the percentage of loci pairs in significant LD. The elevated levels of LD in subgroup two based on loci pairs in significant LD can be due to the higher number of accessions investigated in subgroup two (109) compared to subgroup one (64). A similar observation was made by Malysheva-Otto et al. (2006) in cultivated barley germplasm, where the global collection of 953 barley accessions showed higher intrachromosomal as well as interchromosomal LD (100 and 98%, respectively) than a subset of 207 European 2-rowed spring barley accessions (45 and 42%, respectively). However, subgroup two in our study—even after resampling with 46 accessions (data not shown)—showed higher levels of both interchromosomal and intrachromosomal LD than subgroup one resampled with 46 accessions as well (data not shown). Higher interchromosomal LD for both subgroups showing relatedness may be the cause for LD between SSR loci (see Stich et al. 2006). The r 2 values in our study were comparable to the ones reported for maize and sorghum with SSRs/RFLPs (Hamblin et al. 2004; Remington et al. 2001). In our study, higher mean r 2 values of subgroup one than subgroup two indicate that an increase in LD can occur for accessions within subpopulations with similar genetic background (subgroup one) in contrast to the populations with different genetic backgrounds (subgroup two). This higher LD in subgroup one compared to subgroup two may have been the result of West African farmers’ strong preference to select (Rafalski and Morgante 2004) early-to-intermediate maturing sorghum accessions over late maturing ones. Sorghum accessions selected for earliness thus ensure that important growth stages of the crop are completed before the rainy season is over and the risk of crop failure is minimized.
References
Barnaud A, Trigueros G, McKey D, Joly HI (2008) High outcrossing rates in fields with mixed sorghum landraces: how landraces are maintained? Heredity 101:445–452. doi:10.1038/hdy.2008.77
Bhattramakki D, Dong J, Chhabra AK, Hart GE (2000) An integrated SSR and RFLP linkage map of Sorghum bicolor (L.) Moench. Genome 43:988–1002. doi:10.1139/gen-43-6-988
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Brown AHD (1989) Core collections: a practical approach to genetic resources management. Genome 31:818–824. doi:10.1139/g89-144
Brown SM, Hopkins MS, Mitchell SE, Senior ML, Wang TY, Duncan RR, Gonzalez-Candelas F, Kresovich S (1996) Multiple methods for the identification of polymorphic simple sequence repeats in sorghum [Sorghum bicolor (L.) Moench]. Theor Appl Genet 93:190–198. doi:10.1007/BF00225745
Buckler ES, Thornsberry JM (2002) Plant molecular diversity and applications to genomics. Curr Opin Plant Biol 5:107–111. doi:10.1016/S1369-5266(02)00238-8
Casa AM, Mitchell SE, Hamblin MT, Sun H, Bowers JE, Paterson AH, Aquadro CF, Kresovich S (2005) Diversity and selection in sorghum: simultaneous analyses using simple sequence repeats. Theor Appl Genet 111:23–30. doi:10.1007/s00122-005-1952-5
Casa AM, Pressoir G, Brown PJ, Mitchell SE, Rooney WL, Tuinstra MR, Franks CD, Kresovich S (2008) Community resources and strategies for association mapping in Sorghum. Crop Sci 48:30–40. doi:10.2135/cropsci2007.02.0080
de Vries J, Toenniessen G (2001) Securing the Harvest: biotechnology, breeding and seed systems for African Crops. CABI Publ, Wallingford
de Wet JMJ, Harlan JR, Kurmarohita B (1972) Origin and evolution of Guinea sorghums. East Afr Agric For J 38:114–119
Deu M, Sagnard F, Chantereau J, Calatayud C, Hérault D, Mariac C, Pham JL, Vigouroux Y, Kapran I, Traore PS, Mamadou A, Gerard B, Ndjeunga J, Bezançon G (2008) Niger-wide assessment of in situ sorghum genetic diversity with microsatellite markers. Theor Appl Genet 116:903–913. doi:10.1007/s00122-008-0721-7
Djè Y, Forcioli D, Ater M, Lefèbvre C, Vekemans X (1999) Assessing population genetic structure of sorghum landraces from North-western Morocco using allozyme and microsatellite markers. Theor Appl Genet 99:157–163. doi:10.1007/s001220051220
Djè Y, Heuertz M, Lefèbvre C, Vekemans X (2000) Assessment of genetic diversity within and among germplasm accessions in cultivated sorghum using microsatellite markers. Theor Appl Genet 100:918–925. doi:10.1007/s001220051371
Doggett H (1988) Sorghum. Longman Scientific & Technical, Essex
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
FAOSTAT (2007) Crop production yearbook. FAO, Rome
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Ann Rev Plant Biol 54:357–374. doi:10.1146/annurev.arplant.54.031902.134907
Folkertsma RT, Rattunde FH, Chandra S, Soma Raju G, Hash CT (2005) The pattern of genetic diversity of Guinea-race Sorghum bicolor (L.) Moench landraces as revealed with SSR markers. Theor Appl Genet 111:399–409. doi:10.1007/s00122-005-1949-0
Ghebru B, Schmidt RJ, Bennetzen JL (2002) Genetic diversity of Eritrean sorghum landraces assessed with simple sequence repeat (SSR) markers. Theor Appl Genet 105:229–236. doi:10.1007/s00122-002-0929-x
Goulão L, Cabrita L, Oliveira CM, Leitao JM (2001) Comparing RAPD and AFLPTM analysis in discrimination and estimation of genetic similarities among apple (Malus domestica Borkh.) cultivars. Euphytica 119:259–270. doi:10.1023/A:1017519920447
Grenier C, Deu M, Kresovich S, Bramel-Cox PJ, Hamon P (2000) Assessment of genetic diversity in three subsets constituted from the ICRISAT sorghum collection using random vs. non-random sampling procedures. B. Using molecular markers. Theor Appl Genet 101:197–202. doi:10.1007/s001220051469
Hamblin MT, Mitchell SE, White GM, Gallego J, Kukatla R, Wing R, Paterson AH, Kresovich S (2004) Comparative population genetics of the Panicoid grasses: sequence polymorphism, linkage disequilibrium and selection in a diverse sample of Sorghum bicolor. Genetics 167:471–483
Harlan JR, de Wet JMJ (1972) A simplified classification of cultivated sorghum. Crop Sci 12:172–176. doi:10.2135/cropsci1972.0011183X001200020005x
Ihaka R, Gentleman R (1996) A language for data analysis and graphics. J Comput Graph Stat 3:299–314
Kalinowski ST (2004) Counting alleles with rarefaction: private alleles and hierarchical sampling design. Conserv Genet 5:539–543. doi:10.1023/B:COGE.0000041021.91777.1a
Kong L, Dong J, Hart G (2000) Characteristics, linkage-map positions, and allelic differentiation of Sorghum bicolor (L.) Moench DNA simple sequence repeats (SSRs). Theor Appl Genet 101:438–448. doi:10.1007/s001220051501
Kraakman ATW, Niks RE, Van der Berg PMMM, Stam P, Van Eeuwijk FA (2004) Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 168:435–446. doi:10.1534/genetics.104.026831
Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048. doi:10.1126/science.8091226
Lewis PO, Zaykin D (2002) GDA manual. University of North Carolina Press, Chapel Hill
Malysheva-Otto LV, Ganal MW, Roder MS (2006) Analysis of molecular diversity, population structure and linkage disequilibrium in a worldwide survey of cultivated barley germplasm (Hordeum vulgare L.). BMC Genet 7:1471–2156. doi:10.1186/1471-2156-7-6
Maurer HP, Melchinger AE, Frisch M (2004) PLABSOFT: software for simulation and data analysis in plant breeding. In: 17th EUCARPIA Gen Congr 2004 (poster abstr) Tulln
Morden CW, Doebley J, Schertz KF (1989) Allozyme variation in old world races of Sorghum bicolor (Poaceae). Am J Bot 76:247–255
Morrell PL, Lundy KE, Clegg MT (2003) Distinct geographic patterns of genetic diversity are maintained in wild barley (Hordeum vulgare ssp. spontaneum) despite migration. Proc Natl Acad Sci USA 100:10812–10817. doi:10.1073/pnas.1633708100
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Niangado O (2001) The state of millet diversity and its use in West Africa. In: Cooper HD, Spillane C, Hodgin T (eds) Broadening the genetic base of crop production. IPGRI/FAO, Rome, pp 147–157
Nordborg M (2000) Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154:923–929
Ollitrault O, Noyer JL, Chantereau J, Glaszmann JC (1997) Structure génétique et dynamique des variétiés traditionnelles de sorgho au Burkina Faso. In: Begic A (ed) Gestion des Resources Génétiques de Plantes en Afrique des Savanes. IER-BRG Solagral, Bamako, pp 231–240
Omanya GO (2005) Evaluating ICRISAT’s Breeding Strategy for Pearl Millet with Farmers in West and Central Africa. Final project report for June 2001–November 2004, submitted to Deutsche Gesellschaft für Technische Zusammenarbeit (GTZ) GmbH
Paterson AH, Bowers JE, Bruggmann R (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556. doi:10.1038/nature07723
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Purseglove JW (1985) Tropical crops: monocotyledons. Longman, London
Rafalski A, Morgante M (2004) Corn and Humans: recombination and linkage disequilibrium in two genomes of similar size. Trends Genet 20(2):103–111
Ramu P, Kassahun B, Senthilvel S, Ashok Kumar C, Jayashree B, Folkertsma RT, Ananda Reddy L, Kuruvinashetti MS, Haussmann BIG, Hash CT (2009) Exploiting rice-sorghum synteny for targeted development of EST-SSRs to enrich the sorghum genetic linkage map. Theor Appl Genet 119:1193–1204. doi:10.1007/s00122-009-1120-4
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484. doi:10.1073/pnas.201394398
Rooney WL, Smith CW (2000) Techniques for developing new cultivars. In: Smith CW, Frederiksen RA (eds) Sorghum. Wiley, New York, pp 329–347
Saghai Maroof MA, Solima KM, Jorgenson RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Schloss SJ, Mitchell SE, White GM, Kukatla R, Bowers JE, Paterson AH, Kresovich S (2002) Characterization of RFLP probe sequences for gene discovery and SSR development in Sorghum bicolor (L.) Moench. Theor Appl Genet 105:912–920. doi:10.1007/s00122-002-0991-4
Sham PC, Zhao JH, Curtis D (2000) The effect of marker characteristics on the power to detect linkage disequilibrium due to single or multiple ancestral mutations. Ann Hum Genet 64:161–169. doi:10.1046/j.1469-1809.2000.6420161.x
Smith CW, Frederiksen RA (2000) Sorghum: origin, history, technology, and production. Wiley, New York
Stich B, Melchinger AE, Frisch M, Maurer HP, Heckenberger M, Reif JC (2005) Linkage disequilibrium in European elite maize germplasm investigated with SSRs. Theor Appl Genet 111:723–730. doi:10.1007/s00122-005-2057-x
Stich B, Maurer HP, Melchinger AE, Frisch M, Heckenberger M, van der Voort JR, Peleman J, Sorensen AP, Reif JC (2006) Comparison of linkage disequilibrium in elite European maize inbred lines using AFLP and SSR markers. Mol Breed 17:217–226. doi:10.1007/s11032-005-5296-2
Szalma SJ, Buckler ES, Snook ME, McMullen MD (2005) Association analysis of candidate genes for maysin and chlorogenic acid accumulation in maize silks. Theor Appl Genet 110:1324–1333. doi:10.1007/s00122-005-1973-0
Taramino G, Tarchini R, Ferrario S, Lee M, Pe ME (1997) Characterization and mapping of simple sequence repeats (SSRs) in Sorghum bicolor. Theor Appl Genet 95:66–72. doi:10.1007/s001220050533
Vaksman M, Traoré S, Niangado O (1996) Le photopériodisme des sorghos africains. Agric Dév 9:13–18
Weir BS (1996) Genetic data analysis II, 2nd edn. Sinauer, Sunderland
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160. doi:10.1016/j.copbio.2006.02.003
Acknowledgments
This research was funded by the Federal Ministry for Economic Cooperation and Development, Germany (ICRISAT/GTZ Project No. 05.7860.9-001.00). We thank two anonymous reviewers for their valuable comments.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Bhosale, S.U., Stich, B., Rattunde, H.F.W. et al. Population structure in sorghum accessions from West Africa differing in race and maturity class. Genetica 139, 453–463 (2011). https://doi.org/10.1007/s10709-011-9564-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-011-9564-2