
Abstract
The objectives of the study were to assess genome wide association study (GWAS) for sugarcane on a panel of 183 accessions and to evaluate the impact of population structure and family relatedness on QTL detection. The panel was genotyped with 3327 AFLP, DArT and SSR markers and phenotyped for 13 traits related to agro-morphology, sugar yield, bagasse content and disease resistances. Marker-trait associations were detected using (i) general linear models that took population structure into account with either a Q matrix from STRUCTURE software or principal components from a principal component analysis added as covariates, and (ii) mixed linear models that took into account both population structure and family relatedness estimated using a similarity matrix K* computed using Jaccard’s coefficient. With general linear models analysis, test statistics were inflated in most cases, while mixed linear models analysis allowed the inflation of test statistics to be controlled in most cases. When only detections in which both population structure and family relatedness were correctly controlled were considered, only 11 markers were significantly associated with three out of the 13. Among these 11 markers, six were linked to the major resistance gene Bru1, which has already been identified. Our results confirm that the use of GWAS is feasible for sugarcane in spite of its complex polyploid genome but also underline the need to take into account family relatedness and not only population structure. The small number of significant associations detected suggests that a larger population and/or denser genotyping are required to increase the statistical power of association detection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sugarcane (Saccharum spp.) is a major industrial crop in tropical and subtropical areas. It accounts for about 80 % of world production of sucrose and has become an important source of renewable energy (FAOSTAT 2012). Average sugarcane yield has doubled in the last 50 years thanks to breeding and improved agricultural practices (Gouy et al. 2013a), but still appears to be far from achieving its theoretical potential (Waclawovsky et al. 2010). Sugarcane is a semi perennial grass which has the particularity to accumulate sucrose at high concentrations into its stems. Sugarcane is clonally propagated using stem cuttings and cultivated with one plant crop and several ratoon crops, following each annual harvest. Criteria taken into account by breeders include sucrose yield, ratooning ability, disease resistances and, more recently, quantitative and qualitative fiber content for second-generation production of cellulosic ethanol. The sugarcane breeding process is expensive and time consuming as it involves the creation of from hundreds of thousands to a million F1 progenies each year (Matsuoka et al. 2009), followed by about 15 years of selection. Accurate phenotypic selection in the first stages of selection remains a challenge (Skinner 1971, Kimbeng and Cox 2003). The first stages of selection are applied to full-sib families without or with a limited number of replicates due to the high number of progenies, and environmental plot effects could mask the intrinsic values of genotype. In these conditions, individual genotypic values of most traits are difficult to assess, and when based on a single plant or plot without multi-crop or multi-locations, broad sense heritability is low (Skinner et al. 1987). At these early stages, the best support is family based selection, as family based heritability is relatively high for most traits. At these early stages of breeding programs, it is greatly hoped that marker assisted selection will improve the accuracy of selection. Molecular markers are already used to describe genetic diversity, to understand genome structure, to highlight the genetic basis of physiological, developmental and morphological variation, and to detect the quantitative trait loci (QTL) associated with agronomic traits (Gouy et al. 2013a). As genotyping costs continue to decrease (Prasanna et al. 2013), statistical association between molecular markers and phenotypes has become a widely used strategy to identify loci responsible for genetic variation (Würschum 2012). Once QTL effects are accurately estimated (across populations and environments), marker assisted selection should make it possible to identify elite genotypes early in the breeding program. Ultimately, the usefulness of molecular markers in breeding program will depend on the total cost of the experiment (genotyping and phenotyping of the calibration experiment plus genotyping of the individuals under selection to predict their phenotype) versus savings in time and in money. Marker assisted selection could also enhance response to selection, in particular for traits that are difficult to improve using conventional phenotypic selection. Many QTL studies have been conducted on sugarcane, but most were based on bi-parental progenies (Aitken et al. 2008; Aljanabi et al. 2007; Alwala et al. 2009; Da Silva and Bressiani 2005; Hoarau et al. 2002; Ming et al. 2001; Pastina et al. 2012; Raboin et al. 2001; Nibouche et al. 2012; Costet et al. 2012b). Progenies from bi-parental populations have accumulated a limited number of recombination events. This could result in the detection of QTLs that cover many centiMorgan (cM) and could be located far from the causative gene, leading to erroneous estimation of marker effects (Zhu et al. 2008). The more closely the markers are linked to the QTL underlying the variation of the trait, the more efficient the marker selection. Genome wide association studies, also known as linkage disequilibrium-based studies, use diversity panels, e.g. germplasm or core collections. The collections used in such approaches have accumulated many recombination events from several distinct lineages and consequently enable high-resolution mapping (Nordborg and Tavaré 2002). The collections include large allelic diversity as they usually contain a high proportion of natural variation available for breeding purposes and allow the simultaneous analysis of several traits (Yu and Buckler 2006). However, genome wide association studies must deal with more type I & type II errors QTL studies. Control of type I error is a major concern in genome wide association mapping as false marker-trait associations can arise when population stratification is not taken into account (Pritchard et al. 2000). Population stratification generates covariance among individuals, thereby biasing the estimation of allelic effects (Lander and Kruglyak 1995). On the other hand, if a locus is closely associated with genetic stratification, controlling for population stratification can result in false negatives (type-II errors). Empirical studies have demonstrated that a causative locus can disappear when population stratification is taken into account in the analysis (Andersen et al. 2005; Cai et al. 2013; Zhao et al. 2011). Two parameters are usually considered for population stratification (i) the population structure corresponding to relationships among subpopulations or cluster associated with local adaption or diversifying selection and (ii) the familial relatedness corresponding to the relationship among individuals from recent coancestry (Yu et al. 2006). Population structure and familial relatedness could be inferred using genome wide molecular data. Population structure is often captured using a model-based Bayesian clustering algorithm such as the one developed in the STRUCTURE software (Pritchard et al. 2000) to assign individuals to cluster (the Q matrix) or using principal components coordinates of individuals (PC matrix) of a Principal Component Analysis. “Familial relatedness” is generally estimated using a Kinship matrix (K matrix) to define the degree of genetic covariance between each pair of individuals. General linear model can model population structure by including covariates such as PC or Q matrix as fixed effect. Mixed Linear model use a mixture of fixed effects (using the PC or Q matrix as covariates) and random effects (using the K matrix of pairwise kinship coefficients) to model both population structure and familial relatedness (Yu et al. 2006).
The genetics of current sugarcane cultivars are extremely complex, they have a high polyploid genome that result from their interspecific origin between two polyploid ancestral species. The history of sugarcane breeding is recent because all modern sugarcane cultivars are interspecific hybrids deriving from few crosses performed at the end of the 19th century between the domesticated S. officinarum, a sugar-producing species, and the wild S. spontaneum species. Only a few parental founder accessions were involved in these crosses (Roach 1989). Since then, plant material has been exchanged between sugarcane breeding centers and some important cultivars, such as POJ2878 or NCO310, have been used extensively in crosses and are consequently encountered in the genealogy of many modern cultivars. In this situation one can expect cryptic structuration of the population of modern cultivars. Several studies have assessed the genetic diversity and population structure in sugarcane germplasm. Clear genetic structure was revealed in studies that included individuals belonging to different species or genera (modern cultivars Saccharum spp., S. officinarum, S robustum, S. sinense, S. barberi, S. spontaneum, Miscanthus spp. and/or Erianthus spp.) (Besse et al. 1998; Cordeiro et al. 2003; Tai and Miller 2002). However, both unstructured populations (Jannoo et al. 1999; Lu et al. 1994; Raboin et al. 2008) and structured populations (Selvi et al. 2005; Singh et al. 2013; Wei et al. 2006; Wei et al. 2010) were reported in studies that used panels composed of modern hybrid accessions. The recent breeding history of sugarcane cultivars, associated with the limited number of founders, should be a source of linkage disequilibrium (LD), and the potential of LD-based association studies to identify marker-trait associations has already been highlighted in sugarcane (Jannoo et al. 1999; Raboin et al. 2008). Nevertheless, only a few studies have assessed the ability of association mapping in sugarcane to detect associations between markers and traits including sugarcane yield (Wei et al. 2010) and resistance to smut, to African stalk borer, to pachymetra root rot, to leaf scald, and to Fiji leaf gall (Butterfield 2007; McIntyre et al. 2005; Raboin 2005; Wei et al. 2006,). Finally, although some studies have demonstrated the feasibility of GWAS in several plants through the identification of previously known loci (Yu et al. 2006; Zhao et al. 2007; 2011), this is not yet the case for sugarcane where to date, no detected QTL has been confirmed as true positive.
The objectives of this study were thus to (i) evaluate the impact of population structure on the phenotypic variability on a diversity panel of 183 sugarcane cultivars, and (ii) identify markers associated with 13 morphological, technological, agronomic and disease resistance traits, using an association mapping approach.
Materials and methods
Plant material
The present study was based on a 183 sugarcane accession panel sampled from the REUb panel described by Costet et al. (2012a, b). These accessions were bred in 29 sugarcane breeding centers during the curse of the last century. This panel is a representative sample of sugarcane germplasm cultivated worldwide. The 183 accessions cover a wide range of relatedness, from full-sibs to individuals bred from distinct genealogies (ESM 1).
Field trials and phenotyping
The experimental data used in this study are summarized in supplementary material 2 (ESM2). The panel was phenotyped for 13 agronomic traits: sucrose yield, stalk diameter, stalks number, stalk height, bagasse content, brix of the juice, in vitro neutral detergent fiber (NDF) of bagasse digestibility, flowering rate, incidence of Sugarcane yellow leaf virus (SCYLV: polerovirus causing yellow leaf disease), incidence of Melanaphis sacchari (aphid vector of the SCYLV), infection severity of brown rust (a fungal disease caused by Puccinia melanocephala), infection severity of gumming (a bacterial disease caused by Xanthomonas axonopodis pv. vasculorum) and incidence of smut (a fungal disease caused by Sporisium scitaminea). Five different locations scattered throughout Reunion Island (Indian Ocean) were used for phenotyping: Menciol, Bassin-Martin, La Mare, Vue-Belle and Le Gol. The experimental design was an alpha-lattice with three complete replications, each containing incomplete blocks of 10 accessions. At Menciol experimental station, the elementary plot was composed of three rows four meters in length, with 1.5 meter inter-row spacing, in which 15 cuttings with three buds were planted. The trials in the four other locations are detailed in Gouy et al. (2013b). Yield-related traits and flowering rate were phenotyped at Bassin-Martin, Vue-Belle and La Mare, while in vitro NDF of bagasse digestibility and diseases were phenotyped at Menciol, Bassin-Martin and/or Le Gol (ESM 1). Stalk diameter, number of millable stalks, juice brix, bagasse content, in vitro NDF of bagasse digestibility, SCYLV, rust infection severity and smut incidence were measured as described in Gouy et al. (2013b). Stalk height was the length of the millable stalk. Sucrose yield produced per area was estimated from the fresh biomass of the millable stalk weighed on each plot, and from its sucrose content. The juice ratio was estimated using a 500-g sample of fresh pulp pressed using a hydraulic press. The sucrose content of the resulting juice was estimated using a refractometer. Stalk height and sucrose yield were phenotyped at harvest. Flowering rate was measured at harvest by counting the number of stalks with traces of previous flowering, i.e. presence of a panicle axis. Aphid incidence was scored at Bassin Martin every two weeks for 14 weeks in the 2007–2008 cropping season, for 20 weeks in the 2008–2009 cropping season, and for 24 weeks for the 2009–2010 cropping season, giving a total of 29 counts. At each observation date, in each elementary plot, the lowest green leaf on 20 randomly selected stalks was inspected. A leaf was recorded as being infested when at least one aphid was present on it, and the percentage of infested leaves per plot, i.e. aphid incidence, was computed. Weekly aphid infestation data from the field trial were computed as the percentage of infested leaves and summarized by an area under incidence progress curve (AUIPC) computed separately in each cropping season. Resistance to gumming was evaluated in 2012 at Menciol and Bassin Martin station. The strain of Xanthomonas axonopodis pv. vasculorum 3P 664, isolated at the La Mare experimental station, was grown for 24 h on a plate containing Wilbrink medium. Bacteria were suspended in 0.01 M Tris buffer (pH 7) to obtain a suspension of 109 bacteria/ml. Inoculation was performed using the method described by Rott et al. (2011). Symptoms were recorded on all the stalks six months after inoculation using a symptom severity scale ranging from 0 to 6, where 0 = no symptoms, 1 = one chlorosis line; 2 = more than one chlorosis line, 3 = chlorosis of one or several leaves, 4 = leaf necrosis, 5 = dead stalk.
Statistical analysis of traits
To predict vectors of genetic values (\(\hat{g}\)) used for genome wide association mapping, phenotypic data were analyzed using linear mixed models and generalized linear mixed models. A mixed linear model was used for normally distributed traits as: sucrose yield, stalk diameter, stalk number, stalk height, in vitro NDF digestibility, bagasse content, brix and aphids AUIPC. The model can be written as follows:
where y is the vector of phenotypic observations for each trait, β is a vector of fixed effects related to the experimental design including fixed effects of location, year cycle and replication, b is the vector of random incomplete block effects within each replication ~\({\text{N}}(0,{\text{I}}\sigma_{b}^{2} )\), g is the vector of random effects of genotypes ~\({\text{N}}(0,{\text{I}}\sigma_{g}^{2} )\), gt is the vector of random effects of interaction between genotypes and location or year ~\({\text{N}}(0,{\text{I}}\sigma_{gt}^{2} )\), and e is the vector of residual error of the model ~\({\text{N}}(0,{\text{I}}\sigma_{e}^{2} )\). X, Z 1 , Z 2 and Z 3 are incidence matrices, and I is the identity matrix. These linear mixed models were computed using the lme4 package (Bates et al. 2013) and convergence was checked for each analysis. Broad-sense heritability at the experimental design level and coefficients of genetic variation were calculated for the normally distributed traits according to Gallais (1990). The four disease-related traits and the flowering rate were analyzed with generalized linear mixed models, because of their non-Gaussian distributions. We used the R package MCMCglmm (Hadfield 2010) with Markov chain Monte Carlo (MCMC) routines to fit multi-response generalized linear mixed models. The models used were the standard threshold model with probit link function for both gumming and rust scores, a binomial model with logit link function for the incidence of the Sugarcane yellow leaf virus and flowering rate, and an over-dispersed Poisson model with a log link function for the incidence of smut. Each model was run for 50,000 MCMC simulation iterations. We discarded the first 15,000 cycles as burn-in after checking the stability of posterior values. We checked for convergence of model parameter estimates by inspecting the trace plots of the MCMC iterations and autocorrelation plots. We chose a thinning interval of 10 iterations, which resulted in 3,500 posterior distribution samples of model parameter estimates. Because the variance components of the four diseases traits were transformed in the link function scale, the heritabilities of these traits could not be estimated.
Genotyping
AFLP genotyping was performed using the AFLP® Analysis System I (Invitrogen) according to the manufacturer’s recommendations. Thirty-six primer pair combinations were used. AFLP digestions, ligations and amplifications were performed as described in Hoarau et al. (2001). Fluorescent labeling was used and electrophoresis was performed on a 3130xl Genetic Analyzer (Applied Biosystems). The AFLP fingerprints were analyzed visually using GelCompar II software (Applied Maths BVBA). For SSR analysis, two primer pairs corresponding to mSSCIR4 and mSSCIR164 loci were used (Raboin et al. 2006). Fluorescent labeling and electrophoresis were performed as for AFLP. Whole genome profiling was enriched with DArT markers (Heller-Uszynska et al. 2011). Total DNA was sent to the commercial company Diversity Arrays Technology Pty Ltd (Yarralumla, Australia) for genotyping. The DArT, AFLP and SSR markers were coded as presence/absence. Low or high frequency markers (<0.05 and >0.95) or markers with more than 10 % missing data were removed. A total of 3,327 markers (1406 AFLP, 1892 DArT and 29 SSR) were obtained. We used the marker R12H16_PCR located in the target region of the rust resistance gene Bru1 (Asnaghi et al. 2000; Daugrois et al. 1996; Costet et al. 2012a) as a diagnostic marker of Bru1.
Estimation of population structure and family-based relatedness
Two methods were used to assess the genetic structure of the panel: the Bayesian clustering method implemented in STRUCTURE software, version 2.3.4 (Pritchard et al. 2000), and principal component analysis (PCA). Both methods were applied on a subsample of 820 independent DArT markers, selected from the whole DArT marker dataset. To test for independence between each pair of markers, we used Fisher’s exact test with Bonferroni correction for multiple testing, i.e. a critical P value = 2.80 × 10−8. This subsample was used to ensure homogeneous coverage and avoid over-representation of genomic regions that could bias the analysis (Patterson et al. 2006). Bayesian clustering was performed under the admixture model considering allelic frequencies as independent. No prior population information was used. Allelic frequencies in each of the K clusters (ranging from 1 to 20) were estimated after a burn-in period of 30,000 cycles and 150,000 MCMC iterations. The procedure was repeated 20 times for each K value to assess the stability of each model. We computed the quantity ∆K that allows the detection of the most likely number of clusters K (Evanno et al. 2005), using the online software STRUCTURE HARVESTER (Earl and vonHoldt 2012). The most likely Q matrix was computed under the CLUMPP program to find optimal alignments (Jakobsson and Rosenberg 2007). The most likely Q matrix was computed under the CLUMPP program to find optimal alignments (Jakobsson and Rosenberg 2007). PCA provides a useful description of the genetic variation between genotypes (Price et al. 2006) and can reveal family relatedness (McVean 2009; Patterson et al. 2006). The PCA was computed, using the R package FactoMineR, version 1.14 (Husson et al. 2010), after standardization of marker scoring and setting missing data to zero (Patterson et al. 2006). We tested the significance of the first 100 principal components (PC) using the Tracy-Widom test (Patterson et al. 2006) with the R package EigenCorr, version 0.2 (Lee et al. 2011).
Some GWAS methods (Yu et al. 2006) use a Kinship matrix estimated from marker data, which defines the degree of genetic covariance between pairs of individuals (see below). In this approach, the kinship coefficients are computed from the probability of identity by state between pairs of individuals, adjusted by the probability of identity by state between random individuals. Such a kinship statistic cannot be computed for polyploids like sugarcane genotyped with dominant markers (Hardy and Vekemans 2002). Instead, we used a genetic similarity matrix, K* (Yu et al. 2006). Previous studies have demonstrated that genetic similarity is correlated with the coefficient of parentage based on pedigree data (Lima et al. 2002; Plaschke et al. 1995; Tinker et al. 1993). We computed the similarity matrix K* using the subsample of 820 independent DArT markers defined above. The K* matrix was computed with the DARwin software (Perrier and Jacquemoud-Collet 2006) using Jaccard’s similarity coefficient.
Effect of population structure on phenotype
The effect of population structure on trait variability was assessed on the vector of predicted genetic values (\({\hat{\text{g}}}\)) obtained with the linear mixed models (1).
The effect of population structure was estimated with a linear model written as follows:
where \({\hat{\text{g}}}\) is the vector of predicted genetic values for each trait, β the vector of fixed effects related to population structure, and e the vector of residual error of the model ~\({\text{N}}(0,{\text{I}}\sigma_{e}^{2} )\). Y is the incidence matrix, and I the identity matrix. Two representations of population structure β were used: genotype assignment of the rate of membership to the clusters computed from STRUCTURE software, i.e. the Q-matrix, and the significant principal components (PCs) from the PCA. Linear models were computed using R software (R Core Team 2013).
Genome wide association mapping
General linear models were used to model population structure by including covariates PC or Q matrix as fixed effect. Mixed linear model were used to mix fixed effects (using the PC or Q matrix as covariates) and random effects (using the K matrix of pairwise kinship coefficients) in order to model both population structure and familial relatedness (Yu et al. 2006).
Association tests between markers and the 13 predicted genotypic values were performed using eight statistical models, with or without correction for family based relatedness or population structure (Yu et al. 2006). Four general linear models and four mixed linear models were used. General linear models and mixed linear models were performed using TASSEL software, version 3.0 (Bradbury et al. 2007). The four GLM consisted in a linear model without correction for population structure named NAIVE and three linear models with correction for population structure using either the Q-matrix defined by the software STRUCTURE considering two and seven clusters (named GLM-Q2 and GLM-Q7), or the significant PC of the PCA (GLM-PC) as fixed co-factors. The MLM model consisted in a mixed linear model with the genetic similarity matrix K* specified as the model co-variance matrix but without fixed cofarctor. The three other mixed linear models were used, they include either the Q-matrix (MLM-Q2 or MLM-Q7) for two or seven clusters, or the significant PC of the PCA (MLM-PC) as fixed co-factors. We used the false discovery rate (FDR) approach (Benjamini and Hochberg 1995) to control the genome wide type I error due to multiple testing. For each statistical test, FDR (q-value) was computed using the R package fdrtool (Klaus and Strimmer 2012; Strimmer 2008). Marker-trait associations with a FDR value of 0.10 were deemed significant. Markers significantly linked to the same trait were tested for pairwise independence using a Fisher’s exact test with a 0.05 critical P value and grouped in the same haplotypes if associated by transitivity (i.e., if marker X is associated with marker Y and marker Y is associated with marker Z, then the three markers are grouped in the same haplotype) as described by Raboin et al. (2008). Test statistics are inflated in association studies when the genetic structure is not well modeled, leading to numerous false positives or artifactual QTLs (Clayton et al. 2005; Lander and Schork 1994; Voight and Pritchard 2005). Other biases like sample preparation or genotyping assay procedures may also inflate probabilities (Clayton et al. 2005). Quantile–quantile (Q–Q) plots were drawn for each trait to vizualize if the distribution of P values was inflated with respect to the expected distribution in the case of no genetic association. To measure the inflation of the test statistic, we computed the inflation factor λ (Devlin and Roeder 1999) for each statistical model. When λ ~ 1, there is no inflation in test statistics. According to Price et al. (2010), λ should be lower than 1.05 to avoid detection of spurious associations. In our study, the metric λ was computed from the Fisher F-statistics, according to the quantitative nature of the traits studied, following Yu et al. (2006).
Results
Quantitative analysis of traits
Results of quantitative genetics analysis of the 13 traits used in the present study are summarized in Table 1. For all traits, genotypic variance was significantly (P < 0.01) higher than zero. Broad sense heritabilities (H2) computed only for normally distributed traits were moderate to high, ranging from 0.63 for sucrose yield to 0.89 for both stalk diameter and bagasse content. A broad range of genetic variation was observed with coefficients of genetic variation (CVg) ranging from 5.4 % for brix to 23 % for stalk number.
Genetic structure of the panel of accessions
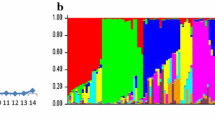
According to Evanno et al. 2005, the ∆K quantity allows the detection of the most likely number of clusters K computed with the Bayesian structuring method implemented in STRUCTURE. We observed the higher values of ∆K for K = 2, K = 5 and K = 7, the latter correspond to the beginning of the plateau of the mean of log likelihoods. The major ∆K value is detected for K = 2 (Fig. 1) suggesting that our panel may originated from the admixture of two populations. Considering K = 2 as the most likely on the basis of a coefficient membership higher than 0.60; we could assign 140 accessions in two clusters. A total of 140 accessions were assigned to a genetic cluster on the basis of a coefficient membership higher than 0.60 (ESM 3). Cluster 1 (C1) comprised 45 accessions, i.e. 24.5 % of the whole panel of 183 accessions. In this genetic cluster, we found accessions bred in 16 different breeding centers with more than half originated from four breeding centers: 22 % from USDA Canal Point in the USA, 16 % from SASRI in South Africa, 11 % from ICAR/SBI Coimbatore in India, and 9 % from FSC Lautoka in Fiji. Cluster 2 (C2) comprised 95 accessions representing 51.9 % of the whole panel. They came from 15 breeding centers. The majority (76 %) of the accessions in C2 originated in four breeding centers: 43 % came from eRcane in Reunion Island, 13 % from HARC in Hawaii, 13 % from MSIRI in Mauritius, and 7 % from WICSBCS Barbados. Accessions originating from seven breeding centers were found in either C1 or C2. No accessions from Hawaii were found in cluster 1, whereas accessions from Hawaii represented 13 % of cluster 2. Accessions from Reunion Island, Mauritius and Barbados accounted for 63 % of cluster 2, while they represented only 11 % of cluster 1. No accessions from Canal Point or Natal (accounting for 38 % of cluster 1) were also found in cluster 2.

Accessions were plotted on the first two principal components (PCs) of the PCA and colored according to their genetic cluster (Fig. 2). The first PC summarizes 4.15 % of total marker inertia. It separates accessions according to the two genetic groups determined by STRUCTURE. The analysis of population structure using PCA revealed that, according to the Tracy-Widom test (P < 0.05), the first 18 PCs were significant.

Principal Component Analysis of 183 sugarcane accessions genotyped with 820 independent DArT markers. Accessions are plotted on the two first axes, PC1 and PC2, the percentage of total inertia represented by each component is in parentheses. Accessions are colored according to their genetic clusters derived from STRUCTURE 2.3.4 analysis. Accessions were assigned to a cluster when they displayed a cluster coefficient membership equal to or higher than 0.60. Accessions belonging to the genetic cluster 1 (C1) are in blue; accessions belonging to the genetic cluster 2 (C2) are in red; accessions not assigned to either genetic clusters are in grey
Impact of the genetic structure of the panel on phenotypic variability
The effect of population structure was assessed on all 13 traits (Table 2). Using assignment in two genetic clusters, i.e. the Q2 matrix, revealed significant effects on seven out of 13 traits. The proportion of variance (R2) explained by cluster assignment ranged from 2.38 % for brix to 15.4 % for stalk diameter. No effects were observed for disease-related traits or flowering rate. The Q7 matrix which corresponds to the STRUCTURE assignment in seven clusters and the first 18 PCs had significant effects on all traits. The proportion of variance explained by the model (R2) ranged from 8.2 % for gumming score to 35.7 % for brix for the model using Q7 and from 16.1 % for rust infection severity to 52.7 % for brix for the model using the18 significant PCs.
Genome wide association mapping
Eight genome wide association models were used to detect marker-trait associations. Population structure was taken into account by including, as covariates, PCs or Q matrix in a general linear model. Mixed Linear model were used in order control both population structure by using the PC or Q matrix as covariates and familial relatedness by using the K matrix of pairwise kinship coefficients (Yu et al. 2006).
Inflation factors λ are summarized in Table 3 and Q–Q plots in ESM 4. For the NAIVE model without control of the genetic structure and familial relatedness of the panel, test statistics were inflated whatever the trait (Fig. 3). Inflation factors λ ranged from 1.31 to 2.59 (Table 3). The GLM-Q2 and the GLM-Q7 models also failed to control the effects of the genetic structure producing consequently inflated probabilities for each trait (Fig. 3, Table 3). For this reason, The NAIVE, GLM-Q2 and GLM-Q7 models were not used for subsequent marker-trait association analysis. The GLM-PC model controlled (λ < 1.05) population structure for five traits out of 13 (Fig. 3, Table 3). Mixed linear models seem to better control inflation of the test statistics with respectively five, seven, seven and nine traits for MLM, MLM-Q2, MLM-Q7 and MLM-PC models (Table 3, Fig. 3).

Example of Quantile–quantile probability plots obtained with four models of genome wide association mapping applied on 13 traits. Models used were a a linear model without correction for population stratification (NAIVE) b a linear model using the Q7-matrix added as a fixed co-factor (GLM-Q7) c a mixed linear model using a similarity matrix specified as the model co-variance matrix (MLM) and, d a mixed linear model using a similarity matrix and the significant eigenvectors from the PCA added as fixed co-factors (MLM-PC). If quantile–quantile probability plots is represented with a + inflation factors ≤1.05, if represented with a dot inflation factors >1.05
The number of significant markers detected for the 13 traits with each model, considering a FDR smaller than 0.10 are summarized in Table 3. As expected from the high inflation of test statistics, the NAIVE and GLM-Q2 models detected numerous significant markers. Depending on the trait, the number of significant markers range from 0 (for aphid AUIPC and In-vitro NDF digestibility) to 526 (for stalk diameter), i.e. 16 % of the whole marker dataset. Using the GLM-Q7 model greatly reduce the number of significant markers (ranging 0 to 23 depending on the trait), even if inflation test statistics is never controlled. In contrast, the five other models which better control the inflation of the test statistics (GLM-PC, MLM, MLM-Q2, MLM-Q7 and MLM-PC) revealed few or no significant markers.
The QTLs detected using the GLM-PC, MLM-PC and MLM-Q models are summarized in Table 4. Considering all traits, a total of 26 significant associations were found at an FDR of 0.10, but only 11 of these markers were detected with a model that shows an inflation factor lower than or equal to 1.05, i.e. models that were assumed to efficiently control the risk of spurious associations. QTL were detected for sucrose yield, brix, in vitro NDF digestibility, flowering rate, rust infection severity and smut incidence. The proportion of total phenotypic variation explained by a single marker range from 6.1 % to 12.5 %. The R2 value obtained with the diagnostic marker of the major rust resistance gene Bru1, R12H16 explain at least 46.3 % of the phenotypic variation. Eight markers were detected for rust infection severity using GLM-PC, three of which were also detected with MLM-PC, two with MLM-Q2 and one with MLM-Q7. Among the markers significantly associated with rust infection severity, the six that had a negative effect were grouped in the same haplotype and four of them were significantly associated with R12H16_PCR. Two markers having positive effects (susceptibility) were not associated with the diagnostic marker R12H16_PCR, but were significantly associated with rust infection severity when the GLM-PC model was used. Two markers were detected for flowering rate using the GLM-PC model. They exhibited positive effects and were independent to each other; one of them was also detected using the MLM-PC model. For sucrose yield, only one marker with a positive effect was detected through the GLM-PC model, but the marker was not detected using MLM. For in vitro NDF digestibility, and smut incidence, associations were observed using the GLM-PC model, or for brix using MLM and MLM-Q2 model, however in these cases inflation factors were always higher than 1.05. For that reason, these associations should be considered with caution.
Discussion
This study provides the first validation of the use of the GWAS strategy in sugarcane as it showed that it is possible to identify a major gene previously identified in biparental progenies. It revealed that association models that include population structure and family-based relatedness can control spurious associations for most sugarcane traits. However in our experimental conditions, only a small number of significant associations were finally detected.
In genome wide association studies, population structure has to be taken into account and modeled correctly as it is the cause of false-positive detections, and consequently leads to a high number of spurious associations (Lander and Schork 1994). We assessed genetic structure using a panel of 183 sugarcane accessions and a Bayesian clustering method implemented in STRUCTURE software (Pritchard et al. 2000) and principal component analysis (PCA). The Bayesian clustering based the method of Evanno et al. 2005 suggest that the most likely number of clusters is two, but small ∆K values are also detected for K = 5 and 7. Using PCA to summarize global genetic variation in the population, we observed no clear structure like that observed in other species including potato (D’hoop et al. 2010), rice (Zhao et al. 2011) and sorghum (Caniato et al. 2011). Both population structure representations, i.e. Bayesian clustering and PCA, explained a significant part of phenotypic variability but we observed differences between the two representations of structure. The most likely Bayesian clustering in two clusters had no significant effects on five traits. The clustering in seven clusters had a significant effect on all traits but whatever the traits the PC from the PCA, which modeled a more complex genetic structure, including part of the family-relatedness (McVean 2009; Patterson et al. 2006), explained higher proportion of the phenotypic variance. The history of sugarcane breeding is recent and the first crosses were limited to a few parental ancestors (Arceneaux 1967). In addition, only a few generations separate modern cultivars from their parental ancestors, thus limiting the number of meiosis. Some important cultivars have been used as progenitors in many breeding programs thus creating relatedness between modern sugarcane cultivars. It has been demonstrated that a population with a small effective size, i.e. that has grown rapidly and recently from a few founders, is subject to cryptic relatedness (Voight and Pritchard 2005). Our results suggest that our panel is affected by cryptic relatedness and population structure which is congruent with the history of sugarcane breeding. Like many populations used for GWAS, our panel belongs in the group IV sample with both population structure and family relationships defined by Zhu et al. (2008).
This genome wide association study revealed 26 significant markers linked to seven traits when FDR was set to 0.10. The significant associations detected for brix, in vitro NDF digestibility, gumming scoring and smut incidence should be considered with caution because of their inflation factor λ, which ranged from 1.14 to 1.29, and which increases the risk of spurious associations. With satisfactory control of the inflation of test statistics (λ < 1.05), 11 markers were significantly associated with three traits out of 13: sucrose yield (1 marker), flowering rate (2 markers) and brown rust infection severity (8 markers). For brown rust, four markers were significantly associated with each other and linked to the major gene Bru1. The two other markers we detected were not statistically correlated with Bru1 and could thus indicate new loci involved in the genetic control of resistance to rust.
Finally, only a few marker-trait associations were detected for the 13 traits analyzed. Wei et al. (2010), who focused on cane yield and sugar content in a population of 480 sugarcane accessions genotyped with 1531 DArT markers,) also found few significant associations. Their study revealed only five significant markers for cane yield and no markers for sugar content were detected (P < 0.0001).
The small number of marker-trait associations detected could be explained by a lack of power of detection in our association study. The power of detection of an association study depends on several factors including population size, the extent of linkage disequilibrium between the marker and the causal locus, which is influenced by the number of markers used, and the effect and frequency of the QTL (Bradbury et al. 2011; Jianbing et al. 2011, Macleod et al. 2010). The highest number of markers (eight) detected was for rust severity. This trait showed favorable conditions for maximizing the power of detection of marker-trait associations with an equivalent proportion of susceptible and resistant accessions, mainly oligogenic genetic determinism and a reliable phenotype, (Costet et al. 2012a). For traits that do not comply with these conditions, our experimental design lacked power. According to Raboin et al. (2008), with the 3,327 polymorphic markers used in the present study and given the high rate of linkage disequilibrium in sugarcane, our coverage should theoretically have been sufficient. However, the study of Grivet and Arruda (2001) demonstrated that the coverage of the genome with molecular markers is not homogenous and that higher coverage can occur in some genomic regions, such as those that came from S. spontaneum parental species. In sugarcane linkage disequilibium appeared to be large, since linkage disequilibium drops only over a distance of 5 cM and instances of linkage disequilibium blocks of 10 to 20 cM are relatively frequent however many blocks in linkage disequilibium may be missed, as the confounding effects of marker dosage due to polyploidy are assumed to mask many instances of linked markers (Costet et al. 2012a). In highly polyploid plants like sugarcane, GWAS can be improved by increasing marker density, by using, for example, recent tools like genotyping-by-sequencing (Elshire et al. 2011). Another reason for the lack of marker detection is correction of the strong effect of population and family-based structure that results in false-negative associations. Previous studies have shown that QTLs tightly linked to the genetic structure may disappear when genetic structure is modeled (Andersen et al. 2005; Cai et al. 2013; Zhao et al. 2011). In our case, the traits for which we detected significant markers are those that are the least correlated with genetic structure (Table 2).
To conclude, we have shown that sugarcane population structure and family-based relatedness have strong effects on the phenotype of traits that are important for breeding. These effects have to be correctly modeled in genome wide association studies to avoid spurious associations. The mixed linear models we used were efficient in controlling inflation of the test statistics due to the effect of structure and family-based relatedness, and we identified several significant associations. These results confirm that GWAS can be used for sugarcane, but underline the need to control family relatedness and not only population structure. Nevertheless and despite the large linkage disequilibrium present in sugarcane, the limited number of significant associations detected in the present study suggests that a larger population and/or a denser genotyping are required to increase the statistical power of association detection.
References
Aitken K, Hermann S, Karno K, Bonnett G, McIntyre L, Jackson P (2008) Genetic control of yield related stalk traits in sugarcane. Theor Appl Genet 117:1191–1203
Aljanabi SM, Parmessur Y, Kross H, Dhayan S, Saumtally S, Ramdoyal K, Dookun-Saumtally A (2007) Identification of a major quantitative trait locus (QTL) for yellow spot (Mycovellosiella koepkei) disease resistance in sugarcane. Mol Breed 19:1–14
Alwala S, Kimbeng C, Veremis J, Gravois K (2009) Identification of molecular markers associated with sugar-related traits in a Saccharum interspecific cross. Euphytica 167:127–142
Andersen J, Schrag T, Melchinger A, Zein I, Lübberstedt T (2005) Validation of Dwarf8 polymorphisms associated with flowering time in elite european inbred lines of maize (Zea mays L.). Theor Appl Genet 111:206–217
Arceneaux G (1967) Cultivated sugarcanes of the world and their botanical derivation. Proc Int Sug Cane Technol 12:844–854
Asnaghi C, Paulet F, Kaye C, Grivet L, Deu M, Glaszmann J, D’Hont A (2000) Application of synteny across Poaceae to determine the map location of a sugarcane rust resistance gene. Theor Appl Genet 101:962–969
Bates D, Maechler M, Bolker B (2013) lme4: Linear mixed-effects models using S4 classes. R package version 0.999999-2. http://CRAN.R-project.org/package=lme4
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Series B (Methodological) 57:289–300
Besse P, Taylor G, Carroll B, Berding N, Burner D, McIntyre C (1998) Assessing genetic diversity in a sugarcane germplasm collection using an automated AFLP analysis. Genetica 104:143–153
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Bradbury P, Parker T, Hamblin MT, Jannink JL (2011) Assessment of power and false discovery rate in genome-wide association studies using the BarleyCAP germplasm. Crop Sci 51:52–59
Butterfield M (2007). Marker assisted breeding in sugarcane: a complex polyploid, University of Stellenbosch. PhD Thesis: 164 pp
Cai S, Wu D, Jabeen Z, Huang Y, Huang Y, Zhang G (2013) Genome-wide association analysis of aluminum tolerance in cultivated and tibetan wild barley. PLoS ONE 8:e69776
Caniato FF, Guimarães CT, Hamblin M, Billot C, Rami J-F, Hufnagel B, Kochian LV, Liu J, Garcia AAF, Hash CT, Ramu P, Mitchell S, Kresovich S, Oliveira AC, de Avellar G, Borém A, Glaszmann J-C, Schaffert RE, Magalhaes JV (2011) The relationship between population structure and aluminum tolerance in cultivated sorghum. PLoS ONE 6:e20830
Clayton DG, Walker NM, Smyth DJ, Pask R, Cooper JD, Maier LM, Smink LJ, Lam AC, Ovington NR, Stevens HE, Nutland S, Howson JMM, Faham M, Moorhead M, Jones HB, Falkowski M, Hardenbol P, Willis TD, Todd JA (2005) Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat Genet 37:1243–1246
Cordeiro GM, Pan Y-B, Henry RJ (2003) Sugarcane microsatellites for the assessment of genetic diversity in sugarcane germplasm. Plant Sci 165:181–189
Costet L, Le Cunff L, Royaert S, Raboin L-M, Hervouet C, Toubi L, Telismart H, Garsmeur O, Rousselle Y, Pauquet J, Nibouche S, Glaszmann J-C, Hoarau J-Y, D’Hont A (2012a) Haplotype structure around Bru1 reveals a narrow genetic basis for brown rust resistance in modern sugarcane cultivars. Theor Appl Genet 125:825–836
Costet L, Raboin L-M, Payet M, D’Hont A, Nibouche S (2012b) A major QTA for resistance to the Sugarcane yellow leaf virus (Luteoviridae). Plant Breed 131:637–640
D’Hoop BB, Paulo MJ, Kowitwanich K, Sengers MI, Visser RGF, Eck HJV, van Eeuwijk F (2010) Population structure and linkage disequilibrium unravelled in tetraploid potato. Theor Appl Genet 121:1151–1170
Da Silva JA, Bressiani JA (2005) Sucrose synthase molecular marker associated with sugar content in elite sugarcane progeny. Genet Mol Biol 28:294–298
Daugrois J, Grivet L, Roques D, Hoarau J, Lombard H, Glaszmann J-C, D’Hont A (1996) A putative major gene for rust resistance linked with a RFLP marker in sugarcane cultivar ‘R570′. Theor Appl Genet 92:1059–1064
Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004
Earl DA, vonHoldt BM (2012) Structure Harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conser Genet Resour 4:359–361
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
FAOSTAT (2012) http://faostat.fao.org/site/567/DesktopDefault.aspx?PageID=567#ancor
Gallais A (1990) Théorie de la sélection en amélioration des plantes, Masson edn. France, Paris
Gouy M, Nibouche S, Hoarau JY, Costet L (2013a) Improvement of yield per se in sugarcane. In: Varshney RK, Tuberosa R (eds) Translational genomics for crop breeding: abiotic stress, yield, and quality. John Wiley & Sons, Inc., Hoboken, pp 211–238
Gouy M, Rousselle Y, Bastianelli D, Lecomte P, Bonnal L, Roques D, Efile J-C, Roche S, Daugrois J, Toubi L, Nabenza S, Hervouet C, Telismart H, Denis M, Thong Chane A, Glaszmann JC, Hoarau J-Y, Nibouche S, Costet L (2013b) Experimental assessment of the accuracy of genomic selection in sugarcane. Theor Appl Genet 126:2575–2586
Grivet L, Arruda P (2001) Sugarcane genomics: depicting the complex genome of an important tropical crop. Cur Opin Plant Biol 5:122–127
Hadfield JD (2010) MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R package. J Stat Softw 33:1–22
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Heller-Uszynska K, Uszynski G, Huttner E, Evers M, Carlig J, Caig V, Aitken K, Jackson P, Piperidis G, Cox M, Gilmour R, D’Hont A, Butterfield M, Glaszmann J-C, Kilian A (2011) Diversity Arrays Technology effectively reveals DNA polymorphism in a large and complex genome of sugarcane. Mol Breed 28:37–55
Hoarau J-Y, Offmann B, D’Hont A, Risterucci A, Glaszmann JC, Roques D, Grivet L (2001) Genetic dissection of a modern sugarcane cultivar (Saccharum spp.). I. Genome mapping with AFLP markers. Theor Appl Genet 103:84–97
Hoarau J-Y, Grivet L, Offmann B, Raboin LM, Diorflar JP, Payet J, Hellmann M, D’Hont A, Glaszmann JC (2002) Genetic dissection of a modern sugarcane cultivar (Saccharum spp.). II. Detection of QTLs for yield components. Theor Appl Genet 105:1027–1037
Husson F, Josse J, Le S, J M (2010) FactoMineR: multivariate exploratory data analysis and data mining with R. R package version 1.14. http://cran.r-project.org/web/packages/FactoMineR/
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jannoo N, Grivet L, Seguin M, Paulet F, Domaingue R, Rao PS, Dookun A, D’Hont A, Glaszmann JC (1999) Molecular investigation of the genetic base of sugarcane cultivars. Theor Appl Genet 99:171–184
Jianbing Y, Warburton M, Crouch J (2011) Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci 51:433–449
Kimbeng CA, Cox MC (2003) Early generation selection of sugarcane families and clones in Australia: a review. J Am Soc Sug Technol 23:21–39
Klaus B and Strimmer K (2012) fdrtool: Estimation of (local) false discovery rates and higher criticism. R package version1.2.10. http://CRAN.R-project.org/package=fdrtool
Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247
Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037
Lee S, Wright FA, Zou F (2011) Control of population stratification by correlation-selected principal components. Biometrics 67:967–974
Lima MLA, Garcia AAF, Oliveira KM, Matsuoka S, Arizono H, de Souza Jr CL, de Souza AP (2002) Analysis of genetic similarity detected by AFLP and coefficient of parentage among genotypes of sugar cane (Saccharum spp.). Theor Appl Genet 104:30–38
Lu Y, D’Hont A, Paulet F, Grivet L, Arnaud M, Glaszmann JC (1994) Molecular diversity and genome structure in modern sugarcane varieties. Euphytica 78:217–226
MacLeod IM, Hayes BJ, Savin KW, Chamberlain AJ, McPartlan HC, Goddard ME (2010) Power of a genome scan to detect and locate quantitative trait loci in cattle using dense single nucleotide polymorphisms. J Anim Breed Genet 127:133–142
Matsuoka S, Ferro J, Arruda P (2009) The Brazilian experience of sugarcane ethanol industry. In Vitro Cell Dev Biol: Plant 45:372–381
McIntyre C, Whan V, Croft B, Magarey R, Smith G (2005) Identification and validation of molecular markers associated with pachymetra root rot and brown rust resistance in sugarcane using map- and association-based approaches. Mol Breed 16:151–161
McVean G (2009) A genealogical interpretation of principal components analysis. PLoS Genet 5:e1000686
Ming R, Liu S-C, Moore PH, Irvine JE, Paterson AH (2001) QTL analysis in a complex autopolyploid: genetic control of sugar content in sugarcane. Genome Res 11:2075–2084
Nibouche S, Raboin LM, Hoarau J-Y, D’Hont A, Costet L (2012) Quantitative trait loci for sugarcane resistance to the spotted stem borer Chilo sacchariphagus. Mol Breed 29:129–135
Nordborg M, Tavaré S (2002) Linkage disequilibrium: what history has to tell us. Trends Genet 18:83–90
Pastina M, Malosetti M, Gazaffi R, Mollinari M, Margarido GRA, Oliveira K, Pinto L, Souza A, van Eeuwijk F, Garcia AAF (2012) A mixed model QTL analysis for sugarcane multiple-harvest-location trial data. Theor Appl Genet 124:835–849
Patterson N, Price AL, Reich D (2006) Population structure and eigenanalysis. PLoS Genet 2:2074–2092
Perrier X, Jacquemoud-Collet J (2006) DARwin software http://darwin.cirad.fr/
Plaschke J, Ganal MW, Roder MS (1995) Detection of genetic diversity in closely related bread wheat using microsatellite markers. Theor Appl Genet 91:1001–1007
Prasanna B, Cairns J, Xu Y (2013) Genomic tools and strategies for breeding climate resilient cereals. In: Kole C (ed) Genomics and breeding for climate-resilient crops, vol 2, p 487, Springer, pp 213–239
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909
Price AL, Zaitlen NA, Reich D, Patterson N (2010) New approaches to population stratification in genome-wide association studies. Nat Rev Genet 11:459–463
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raboin L-M (2005) Génétique de la résistance au charbon de la canne à sucre causé par Ustilago scitaminea: caractérisation de la diversité génétique du pathogène, cartographie de QTL dans un croisement bi-parental et étude d’associations dans une population de cultivars modernes. Thèse de doctorat, Montpellier, France, ENSAM 119p
Raboin L-M, Offmann B, Hoarau J-Y, Notaise J, Costet L, Telismart H, D’Hont A (2001) Undertaking genetic mapping of sugarcane smut resistance. In Proc. S Afr Sug Technol Ass 75:94–98
Raboin L-M, Oliveira K, Lecunff L, Telismart H, Roques D, Butterfield M, Hoarau J, D‘Hont A (2006) Genetic mapping in sugarcane, a high polyploid, using bi-parental progeny: identification of a gene controlling stalk colour and a new rust resistance gene. Theor Appl Genet 112:1382–1391
Raboin L-M, Pauquet J, Butterfield M, D’Hont A, Glaszmann J-C (2008) Analysis of genome-wide linkage disequilibrium in the highly polyploid sugarcane. Theor Appl Genet 116:701–714
Roach B (1989) Origin and improvement of the genetic base of sugarcane Proc. Aust Soc Sug Technol 11:34–47
Rott P, Fleites L, Marlow G, Royer M, Gabriel DW (2011) Identification of new candidate pathogenicity factors in the xylem-invading pathogen Xanthomonas albilineans by transposon mutagenesis. Mol Plant Microbe In 24:594–605
Selvi A, Nair NV, Noyer JL, Singh NK, Balasundaram N, Bansal KC, Koundal KR, Mohapatra T (2005) Genomic constitution and genetic relationship among the tropical and subtropical indian sugarcane cultivars revealed by AFLP. Crop Sci 45:1750–1757
Singh RK, Jena SN, Khan S, Yadav S, Banarjee N, Raghuvanshi S, Bhardwaj V, Dattamajumder SK, Kapur R, Solomon S, Swapna M, Srivastava S, Tyagi AK (2013) Development, cross-species/genera transferability of novel EST-SSR markers and their utility in revealing population structure and genetic diversity in sugarcane. Gene 524:309–329
Skinner J (1971) Selection in sugarcane: a review. Proc Int Soc Sug Technol 14:149–162
Skinner JC, Hogarth DM, Wu KK (1987) Selection methods, criteria and indices. In: Heinz D (ed) Sugar cane improvement through breeding. Elsevier, Amsterdam, pp 409–453
Strimmer K (2008) A unified approach to false discovery rate estimation. BMC Bioinformatics 9:303–316
Tai P, Miller J (2002) Germplasm diversity among four sugarcane species for sugar composition. Crop Sci 4:958–964
R Core Team (2013) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org/
Tinker NA, Fortin MG, Mather DE (1993) Random amplified polymorphic DNA and pedigree relationships in spring barley. Theor Appl Genet 85:976–984
Voight BF, Pritchard JK (2005) Confounding from cryptic relatedness in case-control association studies. PLoS Genet 1:e32
Waclawovsky AJ, Sato PM, Lembke CG, Moore PH, Souza GM (2010) Sugarcane for bioenergy production: an assessment of yield and regulation of sucrose content. Plant Biotech J 8:263–276
Wei X, Jackson P, McIntyre C, Aitken K, Croft B (2006) Associations between DNA markers and resistance to diseases in sugarcane and effects of population substructure. Theor Appl Genet 114:155–164
Wei X, Jackson PA, Hermann S, Kilian A, Heller-Uszynska K, Deomano E (2010) Simultaneously accounting for population structure, genotype by environment interaction, and spatial variation in marker-trait associations in sugarcane. Genome 53:973–981
Würschum T (2012) Mapping QTL for agronomic traits in breeding populations. Theor Appl Genet 25:201–210
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Cur Opin Biotech 17:155–160
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P, Nordborg M (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3:e4
Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nature Communications 2:467
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. The Plant Genome 1:5–20
Acknowledgments
The authors wish to thank T. Dumont, H. Telismart, C. Lallemand, I. Promi, M. Hoarau and R. Tibère for their contributions to field work and phenotypic data acquisition. This study was funded by the eRcane company, by CIRAD (A French research centre working with developing countries to tackle international agricultural and development issues), by an ATP-SEPANG project grant, by the Conseil Régional de la Réunion, by the European Union (European regional development fund—ERDF), by ANR (Agence Nationale de la Recherche) through the Delicas project, grant ANR-08-GENM-001, and by the ANRT (Association Nationale de la Recherche et de la Technologie) through the CIFRE PhD. Grant No.°600/2012 of M. Gouy.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gouy, M., Rousselle, Y., Thong Chane, A. et al. Genome wide association mapping of agro-morphological and disease resistance traits in sugarcane. Euphytica 202, 269–284 (2015). https://doi.org/10.1007/s10681-014-1294-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-014-1294-y