
Abstract
Taxa considered under low International Union for the Conservation of Nature categories of extinction risk often represent cases of concern to conservation biology. Their high relative abundance precludes management of the entire range due to limited economical resources. Therefore, they require a cost-effective management plan. Borderea pyrenaica (Dioscoreaceae), an endemic plant of the Central Pyrenees and pre-Pyrenees, reaches the French side of the Central Pyrenees on its narrow northernmost boundary at Gavarnie (Parc National des Pyrenées, PNP, France), where it is protected as Vulnerable and considered a priority species. We have used nuclear microsatellite population genetic data to design a management strategy for the 11 populations of B. pyrenaica present in this area and to identify Relevant Genetic Units for its Conservation. The 18 SSR loci analysed identified 56 alleles, 24 of which fulfilled the rarity criterion for this set of populations. Genetic structuring of populations and representativity values derived from regression analyses of probabilities of loss of rare alleles together support differentiation of the B. pyrenaica populations into different management units. Estimates derived from G ST values indicate that five populations would adequately represent the 99.9% of the variation relative to most common alleles whereas calculations based on representativity values indicated that these five populations should equate the proportion 2:2:1 from the three different phylogeographical subdivisions of Gavarnie (Western, Eastern-1 and Eastern-2 ranges). This scheme would allow the preservation of 98.21% of the total B. pyrenaica alleles present in Gavarnie, according to the post glacial history of its populations. This conservation genetic approach could be applied to other low-extinction risk categories of extremely rare and subalpine plants in need of regulatory plans in European National Parks and Natural Reserves.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Modern conservation schemes of endangered and threatened taxa usually involve several sources of data including demography, reproductive biology and genetics. Such designs are aimed at providing a comprehensive picture of population dynamics upon which solid management policies should be founded (López-Pujol et al. 2002; Oostermeijer et al. 2003; Rovira et al. 2002). Because the required degree of protection may depend on the species and on its category of extinction risk, a multidisciplinary approach will likely provide the most efficient conservation plans (Domínguez-Lozano et al. 2003; Heywood and Iriondo 2003). While a precautionary policy for Critically Endangered taxa encourages the protection and monitoring of the entire range of the species, this level of protection is usually either non-existent or limited for those taxa listed as Vulnerable or as a lower category of extinction risk. This is mainly because Vulnerable taxa have many populations and individuals. Generally, not all populations can be covered, so they have to be selected and prioritised for both in situ and ex situ conservation purposes in an objective manner. This is particularly true under limited availability of conservation resources, thus requiring the development of more elaborate genetic studies aimed at identifying relevant genetic units for conservation (RGUC) (Ciofi et al. 1999). Those plans should ensure the conservation of acceptable levels of genetic diversity with the minimum economic effort before a population decline occurs.
Borderea pyrenaica Miégeville is a dioecious strictly sexually-reproducing geophyte (García and Antor 1995a; Segarra and Catalán 2005) endemic to the Central Pyrenees and pre-Pyrenees (Spain–France). With some individuals living more than 300 years, B. pyrenaica has one of the longest life-spans reported for herbaceous plants (García and Antor 1995b). Borderea pyrenaica is regarded as a Tertiary relict of the pantropically distributed Dioscoreaceae (Burkill 1960) that adapted to high mountain habitats where it colonises mobile screes mostly above 1,800 m.a.s.l. Some of these adaptations to such unstable habitats deal with the shift from climbing to dwarf habit coupled with the development of creeping stems and inflorescences (Segarra-Moragues and Catalán 2006). Borderea pyrenaica shows a gemmate pollen ornamentation that is very restricted among the Dioscoreaceae (Caddick et al. 1998; Schols et al. 2003, 2005) and is related to the mainly ant-pollination syndrome of the species (García et al. 1995).
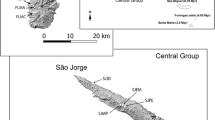
Borderea pyrenaica is restricted to a narrow geographic area of about 160 km2 in the Central Pyrenees and pre-Pyrenees. Most of its populations are located in Spain, where large populations cover wide expanses, whereas in France only 12 populations are known from the Gavarnie cirque (Segarra-Moragues et al. 2007). Historical records from the eastern neighbouring areas at the Troumouse and Estaube cirques (Dupias 1981) have not been confirmed after repeated inspections of potential sites by the Parc National des Pyrénées (PNP) guard service. Despite the short geographical distances between these populations (less than 4 km between the more distant ones; Fig. 1), some geographical discontinuities exist. Three of these populations (Bp01, Bp02 and Bp09) are located in the Western side of the Gavarnie cirque, referred hereafter as WG and are separated from the remaining ones by the central ravine waterfall (Fig. 1). Populations from the Eastern range referred hereafter as EG could be subdivided into two groups according to their separation by a high mountain crest (e.g., EG1: Bp03, Bp04, Bp05, Bp06, Bp10 and Bp11; EG2: Bp07, Bp08 and Bp12; Fig. 1). Population sizes vary according to habitat availability relative to the size of the scree, ranging from more than 5,000 individuals for the largest population of La Planette (Bp01) to only 20 individuals for the smallest population of Chemin du Cirque (Bp03, Table 1), apart from the single female individual found in Belleuve (Bp09). Rough estimates indicate the existence of more than 8,500 individuals in the Gavarnie (PNP) area.

Geographical location of the populations at Gavarnie. Codes of populations correspond to those indicated in Table 1. Triangles, western Gavarnie range; white circles, eastern Gavarnie range, EG1 and grey circles, eastern Gavarnie range, EG2. Bp09 is composed of a single female individual and therefore was not included in this study. Solid line denotes the main geographical discontinuity between WG and EG groups and dotted line denotes the geographical separation between EG1 and EG2 groups, respectively
Because of the narrow occurrence of B. pyrenaica in France, it has been classified under the International Union for the Conservation of Nature (IUCN 2008) category of Vulnerable and included in both the Annex I of the National List of Protected Species in France (Danton and Baffray 1995) and the Red Book of Endangered Flora of France (Olivier et al. 1995). As a consequence, B. pyrenaica has become a priority species within the PNP management plan of endangered Flora. Different studies have been conducted since 1999 in Gavarnie intending to improve the knowledge of B. pyrenaica spatial distribution, its ecology, and the size and fitness of its populations (Valadon and Fallour 2001). Nonetheless, a complementary set of detailed population genetic data are necessary to develop a successful management plan of the species (Valadon 2003). The purpose of out study was to establish the minimum number of conservation units representative of the total genetic variability, taking B. pyrenaica as a model in this microspatial scenario. In this respect, molecular markers have proved useful with respect to the evolutionary and biogeographical processes in plants (Avise 1994; Taberlet et al. 1998; Caujapé-Castells and Jansen 2003), with special relevance at microgeographical scales (Torres et al. 2003a, b; Segarra-Moragues et al. 2005a). Such information has become integral to conservation initiatives of endangered flora (Newton et al. 1999; Segarra-Moragues et al. 2005a, b) because it provides reliable data for comprehensive management and recovery plans without disrupting population relatedness that could have negative evolutionary implications (Prentice et al. 2003).
Earlier molecular studies demonstrated that B. pyrenaica populations were influenced by the oscillatory climatic changes of the Quaternary (Segarra-Moragues and Catalán 2002, 2003). Populations of this taxon were likely sheltered in warmer southern Pyrenean refugia during the cold periods, experiencing a rapid recolonisation after the ice retreat following different pathways to the northernmost areas of Gavarnie where it is currently distributed (Segarra-Moragues et al. 2007). The comparative evolutionary model analysis indicated that the infinite allele model (IAM, Kimura and Crow 1964) explained better than the stepwise mutation model (SMM, Kimura and Ohta 1978) the genotypic relationships among individuals and populations. This is because of the overwhelming influence of migration and genetic drift over mutation in the Quaternary history of the B. pyrenaica populations (Segarra-Moragues and Catalán 2008). The phylogeographical analysis of microsatellite patterns revealed that the populations of Gavarnie originated from two different migration routes, concordant with the West-East geographical separation, and showing significant overall population differentiation F ST = 0.194 (Segarra-Moragues et al. 2007). The populations of the western range were closely related to the western Spanish Pyrenean core of the Ordesa valley whereas those of the eastern range were closer to the eastern Spanish populations of the Pineta valley, showing a strong between-group differentiation F CT = 0.121 (Segarra-Moragues et al. 2007). This indicates that there are at least two groups of B. pyrenaica populations with different origins and genetic composition in Gavarnie.
In this study we have used microsatellite markers to answer the following questions aimed at addressing the most adequate conservation genetic strategy for this taxon: (1) What is the distribution of the genetic diversity between the different geographical population cores of B. pyrenaica in Gavarnie? (2) What should be the number of populations that need to be preserved to represent adequately the genetic diversity present in this area? (3) Which populations should be selected to better represent the detected genetic diversity and therefore should be prioritised for in situ and ex situ conservation initiatives? The number of extant populations at Gavarnie, coupled with the relative inaccessibility of some of them impedes the continuous monitoring of the whole species range in this area thus suiting well the conservation premises stated for Vulnerable taxa.
Materials and methods
Population sampling, DNA extraction and PCR amplification
A total of 565 individuals collected from all 12 extant populations of B. pyrenaica at Gavarnie were sampled for this study (Table 1; Fig. 1). Population Bp09 was not included because it is composed of a single individual. However, we confirmed that it shared allelic relatedness with both populations Bp01 and Bp02 that correspond to the same geographical division (WG). Whenever possible, a ratio of male to female of 1:1 was kept in the sampling scheme and therefore sampling was restricted to flowering individuals. Fresh leaves from all sampled individuals were dried in silica gel and used for DNA isolation. DNA was extracted following the CTAB protocol of Doyle and Doyle (1990) adapted for miniprep extractions. DNA concentration was calculated by comparing to marker VII (Roche) concentration on gel; samples were diluted to a final concentration of ca. 5 ng/μl in Tris–EDTA 0.1× buffer.
Borderea pyrenaica has been reported to be an allotetraploid taxon based on molecular data (Segarra-Moragues et al. 2004; Catalán et al. 2006). Owing to its hybrid origin, most of the microsatellite primer-pairs amplified different alleles derived from one or the other parental genomes present in this species (Catalán et al. 2006). The duplicate disomic allelic segregation permitted unambiguous encoding of genotypic data derived from 11 out of 17 simple sequence repeats (SSR) primer-pairs developed for the genus. The remaining 6 SSR primer-pairs amplified size-overlapping alleles derived from both parental genomes. Because reliable genotype scoring could not be performed in those 6 SSR regions they were not included in the study (Catalán et al. 2006). Amplification protocols for the different microsatellite regions were described elsewhere (Segarra-Moragues et al. 2003, 2004). Successful amplicons were obtained for all samples and their products were run on an ABI 310 automated sequencer (Applied Biosystems). Fragment lengths were assigned with GENESCAN and GENOTYPER software (Applied Biosystems) using ROX-500 as the internal lane standard.
Statistical analysis of microsatellite data
The 11 primer-pairs used in this study scored for 18 microsatellite loci, showing differential banding profiles. Four of the primer-pairs (Bc1169, Bp126, Bp1286 and Bp2214) amplified only one genetic dosage (from one of the putative parental genomes) encoding 4 SSR loci. The remaining seven primer-pairs (Bc1258, Bc1422, Bc1644, Bc166, Bp2256, Bp2290 and Bp2391) amplified both genomic dosages (from both putative parental genomes), encoding 14 SSR loci. Alleles of these loci could be assigned to their correspondent genomic complement and encoded as for conventional diploid taxa (Catalán et al. 2006). These loci were renamed as Bc1258a,b; Bc1422a,b, etc., respectively, to designate each of the two complements.
We estimated isolation by distance between populations of B. pyrenaica in Gavarnie as a factor likely to increase the effect of genetic drift producing differences in allelic frequencies (cf. Segarra-Moragues and Catalán 2008) and thus promoting local adaptation and an increase in number of different genetic units. Borderea pyrenaica is likely to be prone to this effect given the short dispersal distance of both seeds and pollen producing unique genotypic combinations within populations and geographical areas (Moritz 1994). For this purpose we calculated the correlation between log-transformed geographical distances and pairwise linearized F ST values (i.e. F ST/(1 − F ST), Slatkin (1995)) between populations (Rousset 1997). Mantel tests using 1,000 permutations were conducted using NTSYSpc v. 2.11a (Rohlf 2002).
We used two different approaches to identify RGUCs among the B. pyrenaica populations from Gavarnie. One of these approaches entailed the estimators of population structure of Nei (1973). Total genetic diversity present in Gavarnie (H T) was decomposed into the within-population (H S) and between-population (D ST = H T − H S) components, respectively, which were used to calculate the coefficient of population differentiation (G ST = D ST/H T). These values were calculated for each locus for the whole area of B. pyrenaica in Gavarnie and for each geographical subdivision (WG, EG, EG1 and EG2) within the Gavarnie cirque using GENETIX v. 4.04 (Belkhir et al. 2003). G ST values were then used to estimate the number of populations that should be targeted to represent a given proportion of the genetic diversity according to the equation of Ceska et al. (1997), P = 1 − G nST , where n is the number of populations to be sampled to represent a given proportion (P) of the among-population genetic diversity.
Because of the insensitivity of G ST values to rare alleles (Neel and Cummings 2003), we used a second approach that dealt with the associated probabilities of loss of alleles present at low frequencies (rare alleles). We used the expression L = (1 − p)2N of Bengtsson et al. (1995) that estimates the probability that a sample size of (N) populations fails to include an allele with population frequency p (Caujapé-Castells and Pedrola-Monfort 2004). This analysis was carried out with alleles that fulfilled the following criteria of rarity: (1) had an overall frequency lower than 0.50 and (2) were present in less than 50% of the B. pyrenaica populations from Gavarnie. The observed (L o) and the expected (L e) values of the probabilities of loss were calculated. In both L o and L e p, corresponds to the average allele frequency over the populations in which each allele is present. However, for L o, N in the exponential refers to the number of populations where the allele is actually present, whereas in L e, N refers to the total number of populations considered. In other words, in L e, an even average allele frequency is considered across all populations. −LogL o and −LogL e (y-axis) were plotted against the mean frequency of each allele (x-axis) and used for two linear regressions. The quotient between the slope of the expected regression line (based on L e values) and the slope of the observed regression line (based on L o values) estimates the representativity value (R) that indicates the proportion of rare alleles captured by sampling only one population (Bengtsson et al. 1995; Caujapé-Castells and Pedrola-Monfort 2004). Calculations of the probabilities of loss were carried out using TRANSFORMER-3 (Caujapé-Castells and Baccarani-Rosas 2005).
We based our conclusions on how many and which populations should be sampled through these two different approaches because they are based on different premises. First, G ST is mostly influenced by between-population differences in frequencies of most common alleles (Culley et al. 2002) and second, rare alleles represent newly generated variants that may strengthen the species’ capability to withstand environmental changes (Bengtsson et al. 1995). Therefore, a comprehensive estimate of RGUCs based on both complementary methods seems biologically more meaningful.
Results
The 18 SSR loci analysed detected a total of 56 alleles in the 11 B. pyrenaica populations in our study (Table 2). Seven alleles were exclusive to the western range and nine to the eastern range. Five out of the nine eastern alleles were restricted to single populations of EG1 and none was exclusive to EG2. The remaining 40 alleles were shared between different populations of the three geographical divisions (Table 2). Correlation between pairwise log-transformed geographical distances and linearized F ST values (r = 454) revealed significant (P = 0.010) evidence of isolation by distance between populations. Population structure statistics of Nei (1973) detected a higher proportion of total genetic diversity within populations (>80%) than between populations (Table 3). This result was repeatedly recovered when calculations were conducted on the different geographical hierarchies. The highest value of apportionment of the between-population component was obtained for eastern Gavarnie (EG) (D ST = 0.060, 18.75%), suggesting two geographically separated population groups. The highest value of the population differentiation coefficient (G ST) resulted from the consideration of all populations into a single group (Gavarnie G ST = 0.196). Further subdivision into different geographical hierarchies detected minimum G ST values between populations of the western range (G ST = 0.059) and higher among populations of the eastern range (G ST = 0.189). Similar levels of population differentiation were detected among populations of the two eastern groups (EG1, G ST = 0.119; EG2, G ST = 0.120). Estimation of the number of populations to be monitored to represent a 99.9% of the total genetic diversity attributable to most common alleles resulted in 5 (4.24) populations among all populations considered as a single group. When this estimation was carried out separately for each geographical subdivisions, the estimated number of populations within each range were of 2 (2.44) for WG, 4 (4.15) for EG, 3 (3.25) for EG1 and 3 (3.26) for EG2.
Observed and expected probabilities of loss were calculated for 24 alleles that complied with the rarity premises explained in the methods (Tables 2, 4). The quotient between the slopes of the expected (−LogL e) and the observed (−LogL o) regression lines resulted in a representativity value of R = 0.307 for all Gavarnie populations (Fig. 3a), indicating that the sampling of a single population of Gavarnie would roughly account for the 30.7% of the 24 rare alleles present in this area. Independent estimations of representativities for the different groups of populations gave values of R = 1.000 for WG, R = 0.263 for EG, R = 0.266 for EG1 and R = 0.314 for EG2 (Fig. 3b–e). Based on the mean frequencies of rare alleles and their distribution between the different populations (Table 4), the area where each one of these rare alleles holds the lower probability of loss (i.e., the higher probability to be captured by sampling one population at random) was the western range in ten cases and the eastern range in 14 cases, the latter one being separated into EG1 in ten cases and EG2 in four cases. These values indicate that the proportion of populations to be sampled from each geographical subdivision can be expressed as 0.4:0.6 populations from the western-eastern ranges, respectively. The eastern range can be estimated according to the proportion 10:4, which gives a proportion of populations of 0.7:0.3 for EG1 and EG2, respectively. Considering the three geographical subdivisions independently for the relative proportion of populations to be monitored within each area, these values can approximately equal 0.4:0.4:0.2 for WG, EG1 and EG2, respectively.
Discussion
Genetic diversity may be associated with certain fitness traits, enabling a species to counteract environmental changes. It supplies an increased adaptive capability to new habitats or competitive strength, leading ultimately to a higher biological success rate and to its maintenance in space and time (Gautschi et al. 2002; Paschke et al. 2002; Pluess and Stöcklin 2004). Most of the genetic studies addressing conservation issues have been conducted using molecular markers that are assumed to be selectively neutral (Avise 1994; Manel et al. 2005). This has raised the debate of whether such information may reveal the real evolutionary genetic potential of wild populations (Luikart et al. 2003; Holderegger et al. 2006), suggesting that population and landscape genetic studies should focus on genetic variation at quantitative and adaptive trait loci (Merilä and Crnokrak 2001; McKay and Latta 2002; Holderegger et al. 2008). Although this information would always be desirable, it is only currently available for few non-model organisms. It is not the case of many narrow endemics, such as B. pyrenaica, for which whole-genome knowledge is still scarce. Different studies have found a positive correlation between genetic diversity estimates using neutral markers and population fitness (Gautschi et al. 2002; Paschke et al. 2002; Pluess and Stöcklin 2004). These results agree in that the larger the amount of neutral genetic diversity captured by appropriate management schemes, the higher the probability to retain the evolutionary potential of populations through indirect capturing of genetic variation at selectively non-neutral loci (Bataillon et al. 1996). This is especially true when allelic richness and genetic diversity is unevenly distributed among populations as a consequence of isolation by distance (Bataillon et al. 1996), like in B. pyrenaica (Tables 1, 2; Fig. 2). Accordingly, despite the inability of neutral markers to totally reflect the adaptive trends of the B. pyrenaica individuals, our analysis is an appropriate indirect approach to estimate the potential adaptive differences within the species.

Special emphasis has been traditionally devoted to those taxa regarded as Critically Endangered, which usually have low levels of genetic variation. Genetic data have been incorporated into their conservation plans or have been used to improve ongoing conservation initiatives (Petit et al. 1998; Torres et al. 2003a; Segarra-Moragues et al. 2005a, b; Orellana et al. 2009). Critically Endangered taxa often show extremely restricted ranges of distribution, sometimes restricted to a single population (Segarra-Moragues et al. 2005a). Habitat protection by various administrative means becomes a top priority for these taxa (Laguna et al. 2004). These populations are generally easy to handle, and genetic data can help in designing management strategies to avoid inbreeding depression and genetic erosion. Several studies have emphasized the failure of reintroduction programs due to the lack of adequate levels of genetic diversity in germplasm banks established before the availability of genetic data (Calero et al. 1999; Ibáñez et al. 1999; Maunder et al. 2001). Although some of these species may have been already depauperated at the time of collection, inappropriate collection schemes that ignored or did not have genetic data at hand cannot be discarded.
Conversely, Vulnerable taxa usually have wider distribution ranges and more populations and individuals. This can make management of all their wild stocks difficult and expensive. Even when ex-situ strategies are considered these might fail to include a representative amount of the genetic variation (Maunder et al. 2001) or exceed the number of samples needed, resulting in a loss of efficiency and increased costs if genetic data are ignored (Neel and Cummings 2003). These taxa are ideally suited for genetic analyses. Genetic data can provide information on the levels and structure of genetic variation and can help to identify the population genetic relationships and to suggest the best sampling strategies to preserve adequate and representative levels of genetic diversity with minimum expense (Petit et al. 1998; Fraser and Bernatchez 2001; Fay 2003; Orellana et al. 2009). In this sense, population genetic studies of Vulnerable taxa are highly recommended to identify minimum relevant genetic units for conservation (RGUCs, Ciofi et al. 1999), which should be compatible with the maintenance of the evolutionary potential without interfering with the evolutionary history of the populations (Caujapé-Castells and Pedrola-Monfort 2004; Sánchez et al. 2004; Pérez-Collazos et al. 2008). This is particularly true for European taxa that inhabit high mountain areas and that have experienced different histories of population size contraction–expansion during the climatic changes of the Quaternary (Hewitt 1996, 2000; Taberlet et al. 1998; Stehlik 2000; Larena et al. 2002, 2006) that have conditioned their levels of genetic diversity and the phylogeographical relationships between populations (Vargas 2003). The knowledge of the relationships among populations is of primary interest because conservation genetics is biologically more meaningful if it accords with monophyletic groups (Moritz 1994; Soltis and Gitzendanner 1999).
Borderea pyrenaica fits well into this theoretical context. The restricted French range of this species promoted its classification as Vulnerable (IUCN 2008). In spite of the narrow range occupied in the northern Pyrenees (PNP area), the number of populations exceed human and economical resources for complete management. Furthermore, these populations are located in high, almost unaltered, mountain habitats that are unlikely to be threatened by extrinsic factors, making habitat protection a matter of secondary concern for this taxon. However, recent concerns about a rapid global climate warming in the Pyrenean alpine mountain belt could change its perspectives in the near future (Peñuelas and Boada 2003; Solomon et al. 2007). Therefore, genetic information was needed to devise a successful management plan for this species in this area (Valadon 2003).
How many and which populations?
By now a reliable answer to this question can be provided only in the light of genetic information derived from molecular markers. Clustering analyses based on microsatellite data revealed that the populations of B. pyrenaica present in Gavarnie were derived from two different colonising routes followed during their postglacial immigration into this barren narrow geographical area in the Pleistocene (Segarra-Moragues et al. 2007). Accordingly, populations showed significant isolation by distance despite the short geographical distances separating populations (Fig. 2), strong overall population differentiation F ST = 0.194 and strong between-group geographical differentiation F CT = 0.121 (Segarra-Moragues et al. 2007), supporting the idea that they cannot be considered a single conservation unit. The G ST values and the probabilities of loss of rare alleles both highlight the need to separate the B. pyrenaica area in Gavanie into different management units. First we need to identify the minimum number of populations necessary to represent the genetic diversity of the area. G ST values of Gavarnie suggest that the monitoring of five populations would represent the 99.9% of the genetic diversity attributable to most common alleles. This figure exceeds by 9% previous suggestions for the successful accomplishment of conservation goals (Hamrick 1983; Caujapé-Castells and Pedrola-Monfort 2004) but agrees with the values recommended for narrow range endemics (López-Pujol et al. 2004; Sánchez et al. 2004; Pérez-Collazos et al. 2008). Further consideration of independent G ST values for the three geographical areas of Gavarnie suggest an increase in the number of populations to be monitored to eight. This follows from the different levels of genetic diversity and the frequency variance of most common alleles, for which G ST values are particularly sensitive (Caujapé-Castells and Pedrola-Monfort 2004), among populations. Since similar levels of variation would imply that both estimates should converge to a similar value, the estimate of Gavarnie as a whole, should equal the sum of the estimations for the three geographical divisions separately. Generally this concludes in the need to monitor a larger number of populations (Caujapé-Castells and Pedrola-Monfort 2004). In our case, the differences between both estimates are attributable to the variation in the allele frequencies that are shared among most of the populations. Therefore, there is no need to increase this figure by including more populations but to choose five populations among the more genetically diverse ones (Table 1).
The second question deals with which populations should be monitored to achieve an adequate representation of the genetic diversity of the area. Theoretical population genetics advises that populations should be selected in terms of both allelic richness and heterozygosity (Hamrick et al. 1991; Neel and Cummings 2003), because they represent complementary estimates of both the raw amount of genetic variation, as indicated by the number of alleles, and the heterogeneity of populations. Heterozygosity is also less biased by sampling errors and is correlated with general population fitness (Gautschi et al. 2002; Paschke et al. 2002; Pluess and Stöcklin 2004). An earlier population-genetic study conducted on Spanish and French B. pyrenaica populations detected similar levels of heterozygosity among all populations of this taxon from Gavarnie that had similar censuses and a higher allelic richness in those populations of the western range compared to those of the eastern area (Segarra-Moragues et al. 2007, Table 1). Therefore, larger populations should be prioritized for conservation (cf. Martínez-Palacios et al. 1999) in case some genetic diversity may have passed overlooked by small sample sizes. However, this seems unlikely within our broadly sampled study in which we should have a high probability to detect any allele with a frequency higher than 0.0009.
Representativity values derived from the analysis of the probabilities of loss of rare alleles (Table 4; Fig. 3) suggested that five populations should match the proportion of 2:2:1 each from WG, EG1 and EG2 geographical areas, respectively, implying the monitoring of the entire western range and three populations of the eastern range. We therefore recommend the following populations for conservation: Bp01 and Bp02 from the western range; Bp05 and Bp11 from EG1 and, Bp08 from EG2. A management strategy that guarantees the protection of these sets of populations would account for 23 (95.83%) of the 24 rare alleles in Gavarnie and 55 (98.21%) out of the 56 alleles detected in the French range of B. pyrenaica. Only one allele (Bp126-232), exclusive to Bp06 (Table 2), would be missed by this management scheme unless ex situ germplasm stocks are used as genetic reservoirs or it is eventually spread to other populations via gene flow. The populations selected here could serve for restoration or reinforcement if population decline occurs in any of the neighbouring populations of the same geographical division. Translocation of individuals or seeds between populations from either EG1 and EG2 seems feasible given the low genetic differentiation between these two groups. However, this is not recommended between the two main geographical areas of Gavarnie (WG-EG) since they are derived from different Spanish southern Pyrenean ancestral populations. Although outbreeding depression is unlikely given the low genetic distances among all populations of B. pyrenaica, a degree of local adaptation and outbreeding depression can not be ruled out until experimental crosses between individuals of the two geographical ranges are performed and checked for fitness rates. Therefore, we recommend the management of these two areas separately.

Regression analyses of the average allele frequencies (x-axis) of alleles that fulfilled the criteria of rarity stated in the methods with −log probabilities of loss (y-axis). Solid lines with black dots and dashed lines with empty dots represent −log of the observed and expected probabilities of loss, respectively. The number of rare alleles considered (N A) and the representativity values (R) for each geographical subdivision were: a Gavarnie N A = 24, R = 0.3065; b western Gavarnie (WG), N A = 17, R = 1.000; c eastern Gavarnie (EG), N A = 17, R = 0.2626, and EG hierarchy of two additional ranges; d EG1, N A = 16, R = 0.2663 and e EG2, N A = 5, R = 0.3140
Finally, if ex situ strategies are desirable for conservation of seeds in germplasm stocks from the targeted populations an additional advice can be provided in views of the available genetic data (Table 1). The populations of this rare species were determined not to be in Hardy–Weinberg equilibrium (Segarra-Moragues et al. 2007, Table 1), but deviating towards heterozygote deficiency. The significant positive F IS values were explained as the consequence of the preferential mating between spatially close individuals, which favours inbreeding (Segarra-Moragues et al. 2007). This unexpected result for a dioecious taxon was related to the short dispersal of seeds and pollen that produce dense colonies of genetically related individuals and thus produce a strong within-population genetic structure. Accordingly, the genetic patches within sampling sited of B. pyrenaica are much smaller than the predefined populations. This same result was found for he sister taxon B. chouardii (Gaussen) Heslot, that shares similar reproductive traits with B. pyrenaica (Segarra-Moragues et al. 2005a), and is a likely example of a Wahlund effect. From a practical point of view, it would not be advisable to split geographically defined populations into many smaller genetic patches because this would further complicate their management. However, the strong within-population genetic structure cannot be ignored at the time of collection of seeds for ex situ storage, suggesting that this should be carried out from as many as possible spatially separated groups of individuals within populations to guarantee a good representation of all demes.
General conclusions
Narrow endemics that still have many populations are of concern because the protection of their entire range can be both expensive and politically challenging, so more restrictive conservation strategies are sometimes necessary. A balanced combination between the selection of the minimum number of populations needed to succeed in a conservation goal without compromising future genetic consequences by inadequate management is often desired. This study exploits information of microsatellite variation from a practical point of view and provides appropriate recommendations for the adequate management of the populations of a Vulnerable taxon within a narrow geographical area by identifying RGUCs. Following these recommendations the proposed conservation strategy should minimize costs. This strategy should also guarantee the maintenance of representative amounts of genetic variation that can endow enough evolutionary potential for appropriately collected ex situ stocks and to in situ managed populations. This approach taken here should provide valuable theoretical basis for other candidate taxa needed of similar requirements.
References
Avise JC (1994) Molecular markers, natural history and evolution. Chapman and Hall, London
Bataillon TM, David JL, Schoen DJ (1996) Neutral genetic markers and conservation genetics: simulated germplasm collections. Genetics 144:409–417
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2003) GENETIX 4.04, logiciel sous Windows TM pour la génétique des populations, Laboratoire Génome, Populations, Interactions. CNRS UMR 5000, Université Montpellier II, Montpellier
Bengtsson BO, Weibull P, Ghatnekar L (1995) The loss of alleles by sampling: a study of the common outbreeding grass Festuca ovina over three geographical scales. Hereditas 122:221–238. doi:10.1111/j.1601-5223.1995.00221.x
Burkill IH (1960) The organography and the evolution of Dioscoreaceae, the family of yams. Bot J Linn Soc 56:319–412. doi:10.1111/j.1095-8339.1960.tb02508.x
Caddick LR, Furness CA, Stobart KL, Rudall PJ (1998) Microsporogenesis and pollen morphology in Dioscoreales and allied taxa. Grana 37:321–336
Calero C, Ibáñez O, Mayol M, Rosselló JA (1999) Random amplified polymorphic DNA (RAPD) markers detect a single phenotype in Lysimachia minoricensis J.J. Rodr. (Primulaceae), a wild extinct plant. Mol Ecol 8:2133–2136. doi:10.1046/j.1365-294x.1999.00800.x
Catalán P, Segarra-Moragues JG, Palop-Esteban M, Moreno C, González-Candelas F (2006) A bayesian approach for discriminating among alternative inheritance hypotheses in plant polyploids: the allotetraploid origin of genus Borderea (Dioscoreaceae). Genetics 172:1939–1953. doi:10.1534/genetics.105.042788
Caujapé-Castells J, Baccarani-Rosas M (2005) TRANSFORMER-3: a program for the analysis of molecular population genetic data. EXEGEN software and Jardín Botánico Canario “Viera y Clavijo”. Available at: http://exegen.org/es/download.php
Caujapé-Castells J, Jansen RK (2003) The influence of the Miocene Mediterranean desiccation on the geographical expansion and genetic variation of Androcymbium gramineum (Cav.) McBride (Colchicaceae). Mol Ecol 12:1515–1525. doi:10.1046/j.1365-294X.2003.01839.x
Caujapé-Castells J, Pedrola-Monfort J (2004) Designing ex-situ conservation strategies through the assessment of neutral genetic markers: application to the endangered Androcymbium gramineum. Conserv Genet 5:131–144. doi:10.1023/B:COGE.0000029997.59502.88
Ceska JF, Affolter JM, Hamrick JL (1997) Developing a sampling strategy for Baptisia arachnifera based on allozyme diversity. Conserv Biol 11:1133–1139. doi:10.1046/j.1523-1739.1997.95527.x
Ciofi C, Beaumont M, Swingland IR, Bruford MW (1999) Genetic divergence and units for conservation in the Komodo dragon Varanus komodoensis. Proc Roy Soc B Biol Sci 266:2269–2274. doi:10.1098/rspb.1999.0918
Culley TM, Wallace LE, Gengler-Nowak KM, Crawford DJ (2002) A comparison of two methods of calculating G ST, a genetic measure of population differentiation. Am J Bot 89:460–465
Danton P, Baffray H (1995) Inventaire des plantes protégées de France. AFCEV, Yves Rocher, Nathan
Domínguez-Lozano F, Moreno-Saiz JC, Sainz-Ollero H (2003) Rarity and threat relationships in the conservation planning of Iberian flora. Biodivers Conserv 12:1861–1882. doi:10.1023/A:1024110925948
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Dupias G (1981) Fleurs du Parc National. Atlas Floristique 2. Haute Montagne, étage subalpin 1er partie, France
Fay M (2003) Using genetic data to help guide decisions about sampling. In: Smith RD, Dickie JB, Linington SH, Pritchard HW, Probert RJ (eds) Seed conservation. Turning science into practice. Royal Botanic Gardens, Kew, pp 89–96
Fraser DJ, Bernatchez L (2001) Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol Ecol 10:2741–2752. doi:10.1046/j.0962-1083.2001.01411.x
García MB, Antor RJ (1995a) Sex ratio and sexual dimorphism in the dioecious Borderea pyrenaica (Dioscoreaceae). Oecologia 101:59–67. doi:10.1007/BF00328901
García MB, Antor RJ (1995b) Age and size structure in populations of a long-lived dioecious geophyte: Borderea pyrenaica (Dioscoreaceae). Int J Plant Sci 156:236–243. doi:10.1086/297246
García MB, Antor RJ, Espadaler X (1995) Ant pollination of the palaeoendemic dioecious Borderea pyrenaica (Dioscoreaceae). Plant Syst Evol 198:17–27. doi:10.1007/BF00985105
Gautschi B, Widmer A, Joshi J, Koella JC (2002) Increased frequency of scale anomalies and loss of genetic variation in serially bottlenecked populations of the dice snake, Natrix tesellata. Conserv Genet 3:235–245. doi:10.1023/A:1019924514465
Hamrick JL (1983) The distribution of genetic variation within and among natural plant populations. In: Schonewald-Cox CN, Chambers SM, MacBride B, Thomas WL (eds) Genetics and conservation: a reference for managing wild animal and plant populations. Benjamin/Cummings, Menlo Park, pp 335–348
Hamrick JL, Godt MJW, Murawski DA, Loveless MD (1991) Correlations between species traits and allozyme diversity: implications for conservation biology. In: Falk DA, Holsinger KE (eds) Genetics and conservation of rare plants. Oxford University Press, New York, pp 76–86
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276. doi:10.1111/j.1095-8312.1996.tb01434.x
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913. doi:10.1038/35016000
Heywood VH, Iriondo JM (2003) Plant conservation: old problems, new perspectives. Biol Conserv 113:321–335. doi:10.1016/S0006-3207(03)00121-6
Holderegger R, Kamm U, Gugerli F (2006) Adaptive vs. neutral genetic diversity: implications for landscape genetics. Landsc Ecol 21:797–807. doi:10.1007/s10980-005-5245-9
Holderegger R, Herrmann D, Poncet B, Gugerli F, Thuiller W, Taberlet P, Gielly L, Rioux D, Thuiller W, Brodbeck S, Aubert S, Manel S (2008) Land ahead: using genome scans to identify molecular markers of adaptive relevance. Plant Ecol Divers 1:273–283. doi:10.1080/17550870802338420
Ibáñez O, Calero C, Mayol M, Rosselló JA (1999) Isozyme uniformity in a wild extinct insular plant, Lysimachia minoricensis J.J. Rodr. (Primulaceae). Mol Ecol 8:813–817. doi:10.1046/j.1365-294X.1999.00633.x
IUCN (2008) Red list-categories and criteria (version 3.1). Available at: http://www.iucnredlist.org/
Kimura M, Crow JF (1964) The number of alleles that can be maintained in a finite population. Genetics 49:725–738
Kimura M, Ohta T (1978) Stepwise mutation model and distribution of allelic frequencies in a finite population. Proc Natl Acad Sci USA 75:2868–2872
Laguna E, Deltoro VI, Pèrez-Botella J, Pèrez-Rovira P, Serra LL, Olivares A, Fabregat C (2004) The role of small reserves in plant conservation in a region of high diversity in eastern Spain. Biol Conserv 119:421–426. doi:10.1016/j.biocon.2004.01.001
Larena BG, Aguilar JF, Feliner GN (2002) Glacial-induced altitudinal migrations in Armeria (Plumbaginaceae) inferred from patterns of chloroplast DNA haplotype sharing. Mol Ecol 11:1965–1974. doi:10.1046/j.1365-294X.2002.01594.x
Larena BG, Aguilar JF, Feliner GN (2006) Dispersal across Southern Iberian refugia? Integrating RAPDs, sequence data and morphometrics in Armeria (Plumbaginaceae). Folia Geobot 41:305–322. doi:10.1007/BF02904944
López-Pujol J, Bosch M, Simon J, Blanchè C (2002) Allozyme variation and population structure of the very narrow endemic Seseli farrenyi (Apiaceae). Bot J Linn Soc 138:305–314. doi:10.1046/j.1095-8339.2002.00021.x
López-Pujol J, Bosch M, Simon J, Blanché C (2004) Allozyme diversity in the tetraploid endemic Thymus loscosii (Lamiaceae). Ann Bot 93:323–332. doi:10.1093/aob/mch039
Luikart G, England PR, Jordan S, Taberlet P (2003) The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet 4:981–994. doi:10.1038/nrg1226
Manel S, Gaggiotti OE, Waples RS (2005) Assignment methods: matching biological questions with appropriate techniques. Trends Ecol Evol 20:136–142. doi:10.1016/j.tree.2004.12.004
Martínez-Palacios A, Eguiarte LE, Furnier GR (1999) Genetic diversity of the endangered endemic Agave victoriae-reginae (Agavaceae) in the Chihuahuan desert. Am J Bot 86:1093–1098
Maunder M, Cowan RS, Stranc P, Fay MF (2001) The genetic status and conservation management of two cultivated bulb species extinct in the wild: Tecophilaea cyanocrocus (Chile) and Tulipa sprengeri (Turkey). Conserv Genet 2:193–201. doi:10.1023/A:1012281827757
McKay JK, Latta RG (2002) Adaptive population divergence: markers, QTL and traits. Trends Ecol Evol 17:285–291. doi:10.1016/S0169-5347(02)02478-3
Merilä J, Crnokrak P (2001) Comparison of genetic differentiation at marker loci and quantitative traits. J Evol Biol 14:892–903. doi:10.1046/j.1420-9101.2001.00348.x
Moritz C (1994) Defining “evolutionary significant units” for conservation. Trends Ecol Evol 9:373–375. doi:10.1016/0169-5347(94)90057-4
Neel MC, Cummings MP (2003) Effectiveness of conservation targets in capturing genetic diversity. Conserv Biol 17:219–229. doi:10.1046/j.1523-1739.2003.01352.x
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Newton AC, Allnut TR, Gillies ACM, Lowe AJ, Ennos RA (1999) Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol Evol 14:140–145. doi:10.1016/S0169-5347(98)01555-9
Olivier L, Galland JP, Maurin H (1995) Livre Rouge de la flore menacée de France, Tome 1: espèces prioritaires. MNHN SPN, Collection Patrimoine Naturel 20, Paris
Oostermeijer JGB, Luitjen SH, den Nijs JCM (2003) Integrating demographic and genetic approaches in plant conservation. Biol Conserv 113:389–398. doi:10.1016/S0006-3207(03)00127-7
Orellana MR, López-Pujol J, Blanché C, Rovira AM, Bosch M (2009) Genetic diversity in Delphinium staphisagria (Ranunculaceae), a rare Mediterranean dysploid larkspur with medicinal uses. Genetica 135:221–232. doi:10.1007/s10709-008-9271-9
Paschke M, Abs C, Schmid B (2002) Relationship between population size, allozyme variation, and plant performance in the narrow endemic Cochlearia bavarica. Conserv Genet 3:131–144. doi:10.1023/A:1015293530776
Peñuelas J, Boada M (2003) A global change-induced biome shift in the Montseny mountains (NE Spain). Glob Chang Biol 9:131–140. doi:10.1046/j.1365-2486.2003.00566.x
Pérez-Collazos E, Segarra-Moragues JG, Catalán P (2008) Two approaches for the selection of Relevant Genetics Units for Conservation (RGUCs) in the narrow European endemic steppe plant Boleum asperum (Brassicaceae). Biol J Linn Soc 94:341–354. doi:10.1111/j.1095-8312.2008.00961.x
Petit RJ, El Mousadik A, Pons O (1998) Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12:844–855. doi:10.1111/j.1523-1739.1998.96489.x
Pluess AR, Stöcklin J (2004) Genetic diversity and fitness in Scabiosa columbaria in the Swiss Jura in relation to population size. Conserv Genet 5:145–156. doi:10.1023/B:COGE.0000029999.10808.c2
Prentice HC, Malm JU, Mateu-Andrés I, Segarra-Moragues JG (2003) Allozyme and chloroplast DNA variation in island and mainland populations of the rare Spanish endemic, Silene hifacensis (Caryophyllaceae). Conserv Genet 4:543–555. doi:10.1023/A:1025603328704
Rohlf FJ (2002) NtSYSpc, numerical taxonomy and multivariate analysis system. Version 2.11a, User guide. Exeter software, New York
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rovira AM, Bosch M, Molero J, Blanché C (2002) Pollination ecology and breeding system of the very narrow coastal endemic Seseli farreny (Apiaceae). Effects of population fragmentation. Nord J Bot 22:727–740. doi:10.1111/j.1756-1051.2002.tb01936.x
Sánchez JL, Reyes-Betancort JA, Scholz S, Caujapé-Castells J (2004) Patrones de variación genética poblacional en el endemismo canario Matthiola bolleana Webb ex Christ. Bot Macarones 25:3–13
Schols P, Furness CA, Wilkin P, Smets E, Cielen V, Huysmans S (2003) Pollen morphology of Dioscorea (Dioscoreaceae) and its relation to systematics. Bot J Linn Soc 143:375–390. doi:10.1111/j.1095-8339.2003.00227.x
Schols P, Wilkin P, Furness CA, Huysmans S, Smets E (2005) Pollen evolution in yams (Dioscorea: Dioscoreaceae). Syst Bot 30:750–758. doi:10.1600/036364405775097743
Segarra JG, Catalán P (2005) Borderea Miègeville. In: Aedo C, Herrero A (eds) Flora Iberica XXI. CSIC, Madrid, pp 11–14
Segarra-Moragues JG, Catalán P (2002) Low allozyme variability in the critically endangered Borderea chouardii and in its congener Borderea pyrenaica (Dioscoreaceae), two palaeoendemic relicts from the central Pyrenees. Int J Plant Sci 163:159–166. doi:1058-5893/2002/16301-0015
Segarra-Moragues JG, Catalán P (2003) Life history variation between species of the relictual genus Borderea (Dioscoreaceae): phylogeography, genetic diversity, and population genetic structure assessed by RAPD markers. Biol J Linn Soc 80:483–498. doi:10.1046/j.1095-8312.2003.00247.x
Segarra-Moragues JG, Catalán P (2006) Species boundaries and population divergence in the Pyrenean endemic relict genus Borderea (Dioscoreaceae) as revealed by microsatellite (SSR) and other hypervariable markers. Aliso 22:541–555
Segarra-Moragues JG, Catalán P (2008) Glacial survival, phylogeography, and a comparison of microsatellite evolution models for explaining population structure in two species of dwarf yams (Borderea, Dioscoreaceae) endemic to the central Pyrenees. Plant Ecol Div 1:229–243. doi:10.1080/17550870802349757
Segarra-Moragues JG, Palop-Esteban M, González-Candelas F, Catalán P (2003) Characterization of ten trinucleotide microsatellite loci in the critically endangered Pyrenean yam Borderea chouardii (Dioscoreaceae). Mol Ecol Notes 3:265–267. doi:10.1046/j.1471-8286.2003.00422.x
Segarra-Moragues JG, Palop-Esteban M, González-Candelas F, Catalán P (2004) Characterization of seven (CTT)n microsatellite loci in the Pyrenean endemic Borderea pyrenaica (Dioscoreaceae). Remarks on ploidy level and hybrid origin assessed through allozymes and microsatellite analyses. J Hered 95:177–183. doi:10.1093/jhered/esh028
Segarra-Moragues JG, Palop-Esteban M, González-Candelas F, Catalán P (2005a) On the verge of extinction: genetics of the Critically Endangered Iberian plant species, Borderea chouardii (Dioscoreaceae) and implications for conservation management. Mol Ecol 14:969–982. doi:10.1111/j.1365-294X.2005.02482.x
Segarra-Moragues JG, Iriondo JM, Catalán P (2005b) Genetic typing of germplasm accessions as an aid for the species conservation: the case study of Borderea chouardii (Dioscoreaceae), one of the most Critically Endangered Iberian plants. Ann Bot 96:1283–1292. doi:10.1093/aob/mci280
Segarra-Moragues JG, Palop-Esteban M, González-Candelas F, Catalán P (2007) Nunatak survival vs. tabula rasa in the Central Pyrenees: a study on the endemic plant species Borderea pyrenaica (Dioscoreaceae). J Biogeogr 34:1893–1906. doi:10.1111/j.1365-2699.2007.01740.x
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Solomon S, Qin D, Manning M, Chen Z, Marquis M, Averyt KB, Tignor M, Miller HL (2007) Climate change 2007: the physical science basis. Contribution of working group I to the fourth assessment report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge
Soltis PS, Gitzendanner MA (1999) Molecular systematics and the conservation of rare species. Conserv Biol 13:471–483. doi:10.1046/j.1523-1739.1999.97286.x
Stehlik I (2000) Nunatak and peripheral refugia for alpine plants during Quaternary glaciation in the middle parts of the Alps. Bot Helv 110:25–30
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464. doi:10.1046/j.1365-294x.1998.00289.x
Torres E, Iriondo JM, Pérez C (2003a) Genetic structure of an endangered plant, Antirrhinum microphyllum (Scrophulariaceae): allozyme and RAPD analysis. Am J Bot 90:85–92
Torres E, Iriondo JM, Escudero A, Pérez C (2003b) Analysis of within-population spatial genetic structure in Antirrhinum microphyllum (Scrophulariaceae). Am J Bot 90:1688–1695
Valadon A (2003) Le plan d’action flore vasculaire du Parc National des Pyrenees: strategie, methodes et premiers resultats. Acta Bot Barc 49:127–146
Valadon A, Fallour D (2001) Inventaire d’espèces rares et menacées de la Flore du Parc National des Pyrénées (65). Rapport final Projet FEOGA 030462, PNP, 66 p. plus annexes
Vargas P (2003) Molecular evidence for multiple diversification patterns of alpine plants in Mediterranean Europe. Taxon 52:463–476
Acknowledgments
We thank A. Valadon and D. Fallour-Rubio, members of the scientific staff at the PNP, for launching the conservation genetic strategy of Borderea pyrenaica in Gavarnie, J. Caujapé-Castells for kindly providing the calculation sheet for TRANSFORMER and for his statistical advise. D. Fallour-Rubio, J. V. Andrés, R. Andrés and the forest guards at the National Park of the Pyrenees for sampling facilities. The valuable comments of Andrew Alverson and three anonymous referees improved the quality of the final manuscript. This work has been supported by the Spanish Aragón Government grant project P105/99-AV, the French Parc National des Pyrénées (PNP) grant project 2001-64S and the Fundación BBVA grant project BIOCON05/093. J. G. Segarra-Moragues was funded by a Fundación Aragonesa para la Investigación y Desarrollo (ARAID) postdoctoral contract.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Segarra-Moragues, J.G., Catalán, P. The fewer and the better: prioritization of populations for conservation under limited resources, a genetic study with Borderea pyrenaica (Dioscoreaceae) in the Pyrenean National Park. Genetica 138, 363–376 (2010). https://doi.org/10.1007/s10709-009-9427-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-009-9427-2