
Abstract
We developed and characterized microsatellite markers for the genus Lotus, a large genus of leguminous plants containing many endemic species of conservation interest. The marker system was then used to survey patterns of population genetic variation of Lotus sessilifolius, a Canary Island endemic occurring on four islands (La Palma, El Hierro, La Gomera and Tenerife) with the aim of determining whether any of its populations are worthy of special conservation because of genetic distinctiveness. We found strong differentiation between populations with conspicuous geographical signal revealed by population clustering. Generally, populations from each island grouped together. A very striking exception to this pattern is a single population from Tenerife (Tejina-Milán: Anaga Peninsula), which is separated from other Tenerife populations by both genetic clustering and a STRUCTURE analysis, and also shows signs of inbreeding. The genetic distinctiveness of this population deserves especial conservation attention, and may be related to the ancient geological history of the Anaga Peninsula. Importantly, this investigation sets the stage for conservation genetics research in other highly threatened species in the same genus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Macaronesian Lotus L. (Fabaceae: Loteae) is a group of herbaceous legumes currently comprising some 40 species distributed in the 5 oceanic archipelagos of this region (Canary Islands, the Azores, the Cape Verde islands, Madeira and Selvagens), and of Morocco (Sandral et al. 2006). The group colonized Macaronesia from an ancestor of mainland Africa, and has radiated into a wide variety of habitats from sea level (lowland scrub) up to 2000 m (high Canarian mountains) (Allan et al. 2004).
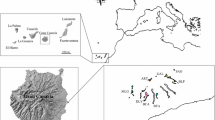
Contrasting with many other congeneric Canarian endemics, Lotus sessilifolius is unusual in its widespread distribution, currently encompassing four islands in the Canary Island archipelago: Tenerife, La Palma, El Hierro and La Gomera (Sandral et al. 2006). This species is currently subdivided into two subspecies. L. sessilifolius DC. subsp. villossisimus (Pitard) Sandral and Sokoloff is exclusively distributed in El Hierro, while L. sessilifolius DC. subsp. sessilifolius is found in Tenerife, La Palma and La Gomera (see Fig. 1). In all the islands, this species is distributed in lowland scrub habitats from sea level to ca. 500 m. L. sessilifolius produces hermaphrodite autogamous yellow flowers adapted to insect pollination (Bombus canariensis Pérez). Fruits are commonly found in all the populations we investigated, and the plants grown under nursery conditions produced viable seeds even in the absence of pollinators. Furthermore, this taxon is of particular interest in that it has been shown to be a close relative of the apparently bird-pollinated Lotus species on the Canary Islands: the Rhyncholotus group (Ojeda et al. 2012).

Map showing the distribution of Lotus sessilifolius in the Canary Island archipelago (square) with the four westernmost islands enlarged. Numbers indicate the approximate sites where each population of L. sessilifolius was collected. Lotus sessilifolius ssp. sessilifolius: 1 = Barranco de Ruiz (Tenerife), 2 = Tejina-Milán (Tenerife), 3 = Güimar (Tenerife), 4 = Poris de Abona (Tenerife), 5 = Mazo (La Palma), 6 = Puntallana (La Gomera); L. sessilifolius ssp. villosissimus: 7 = Punta de Arenas Blancas (El Hierro), 8 = Tecorone (El Hierro)
Microsatellite markers have been widely used to study the genetics of plant populations (Ashley 2010; Kramer et al. 2011), and they have given very important information in a wide variety of Canarian endemic lineages (Sosa et al. 2010; García-Verdugo et al. 2015; Mairal et al. 2015). Our main objectives in this study are (1) to develop nuclear microsatellite markers (SSR) for this particular species and (2) to apply these SSR markers to investigate the intraspecific genetic variation in L. sessilifolius. Although it is a widespread taxon, it consists of many discrete populations fragmented by the separation between islands, and by topographic features within islands. Thus, we also wish to determine whether any of these populations are particularly distinctive at the genetic level and worthy of especial conservation status. These markers will also be useful for studies of other Lotus species in the Canary Islands.
Materials and methods
Study species and DNA isolation
We carried out comprehensive sampling of a key taxon of Macaronesian Lotus: L. sessilifolius DC., throughout its range of distribution. Leaf material was collected from all four islands in the Canary Island archipelago in which it occurs: La Palma, Tenerife, La Gomera and El Hierro (see Fig. 1) and preserved in silica gel. Six individuals per population and a total of eight populations were sampled. Voucher specimens are deposited in the herbarium of the University of British Columbia (UBC). The geographical location of these eight populations is shown in Table 1. Total genomic DNA was extracted using a modified version of the CTAB method (Doyle and Doyle 1987).
Microsatellite selection and amplification
Lotus sessilifolius microsatellites were developed by cross-species amplification of primers designed from Lotus japonicus sequences (Sato et al. 2008), from which we selected 37 microsatellites. Further information about the sequences in relation to the whole genome sequence of L. japonicus is given in Sato et al. (2008). We designed primers in conserved flanking regions of these microsatellites using Primer3 software (Rozen and Skaletsky 2000). Of these, 11 amplified successfully in L. sessilifolius. PCR amplifications were performed in a final volume of 15 μl, containing 10 ng of DNA, 1× PCR buffer (10 mM Tris–HCl pH 8.3, 50 mM KCl), 2.5 mM of each dNTP, 1.5 mM MgCl2, 1 U of Taq DNA polymerase, 0.05 μM forward primer, 0.5 μM reverse primer, and 0.5 μM dye-labeled universal primer. For all 11 loci, the forward primer was synthesized with the universal M13 sequence: TGTAAAACGACGGCCAGT. Four dyes were used VIC, FAM, NED and PET (Applied biosystems, Carlsbad CA, USA) for multiplexing. PCR reactions were performed using a BIO-RAD thermocycler with a touch-down program: 95 °C for 3 min, followed by 9 cycles of 94 °C for 30 s, 65 °C (decreased by 1 °C per cycle) for 30 s and 72 °C for 45 s, followed by 29 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s, and finally followed by a final extension at 72 °C for 20 min. Each microsatellite primer pair was amplified singly and then pooled together based on differences in fluorescent labeling and expected fragment size. An ABI 3730 automated DNA Sequencer (Applied Biosystems) was used to genotype the multiplex sets. The software GeneMapper v.3.2 (Applied Biosystems) was used to score the allele sizes.
Microsatellite data analysis
Genetic variation within L. sessilifolius was determined using the 11 polymorphic microsatellite loci with a total of 48 individuals representing all main populations and taxonomic subdivisions. Due to L. sessilifolius being a tetraploid species, we used computer programs specifically designed for polyploidy analyses. Pairwise genetic distances between all samples were calculated using the R package software POLYSAT (Clark and Jasieniuk 2011). Two genetic distance measures were calculated: Lynch distance (Lynch 1990) and Bruvo distance (Bruvo et al. 2004). Lynch’s distance is based on band-sharing dissimilarity between individuals, and Bruvo’s distance takes into account mutations (Clark and Jasieniuk 2011). Both the Lynch’s and Bruvo’s genetic distance measures showed very similar genetic relationships among the 48 individuals. We used the Lynch’s distance neighbor joining tree to show the individual-based intraspecific genetic relationships. The APE software (Paradis et al. 2004) was used to construct a neighbor joining tree of the 48 individuals based on Lynch distance.
We calculated population genetics statistics using the software AUTOTET (Thrall and Young 2000). For each of the eight populations we calculated allelic richness (A), observed (H O ) and expected heterozygosity (H E ) and inbreeding coefficient (F IS ). The values of H E and F IS were calculated based on random chromosomal segregation (Thrall and Young 2000).
Population allele frequencies were calculated using the ‘simpleFreq’ method in POLYSAT, which assumes all alleles have the same chance of being present in more than one copy (Clark and Jasieniuk 2011). Then these allele frequencies were used to estimate pairwise genetic distances (F ST ) among the eight populations of L. sessilifolius. The APE software (Paradis et al. 2004) was used to construct population-based trees using various algorithms (UPGMA and neighbour-joining) and various population distance metrics (Nei’s D, G ST , and F ST ). UPGMA and neighbour-joining (NJ) are complementary: UPGMA assumes clock-like behaviour of microsatellite variation whereas NJ does not, but UPGMA is very robust when evolutionary rates are high, as in microsatellites (Huelsenbeck and Kirkpatrick 1996). Various measures have been used to compute genetic distances between populations, such as the commonly used F ST , Nei’s standard genetic distance (D), and “unbiased genetic distance” (G ST ) (Nei 1972; Goldstein et al. 1995; Petren et al. 1999; Ochieng et al. 2007).
Population genetic structure was also calculated using the software STRUCTURE 2.3.4 (Pritchard et al. 2000). STRUCTURE is complementary to population clustering. It is not appropriate for inferring the genetic relatedness of distinct populations but can be used for the assignment of individuals to populations and for inferring the existence of distinct populations (Pritchard et al. 2000). We performed 20 replicate runs for all K values using the admixture model and correlated allele frequencies among populations. We used a length of 500,000 burn-in period and 750,000 Markov chain Monte Carlo repetitions, and K values ranging from 1 to 8. The best inference of K was determined using the method of Evanno et al. (Evanno et al. 2005) based on the maximized second-order rate of change in posterior probability for a given K (i.e. ΔK).
Results
Microsatellite variation
Overall, the microsatellite loci were highly polymorphic in the 48 individuals of L. sessilifolius sampled. The number of alleles per locus ranged from two alleles to 21 alleles with an average of 10.82 alleles per locus (Table 2). The patterns of microsatellite alleles per locus per individual were consistent with the tetraploid nature of L. sessilifolius, ranging from one to four.
Population genetic diversity
Population level genetic diversity estimates were similar across the eight populations except for the population Tejina-Milán in Tenerife (pop C), which had the lowest genetic diversity values (Table 3). This population had an allelic richness (A) of 1.09, observed heterozygosity of 0.05 and had the highest inbreeding coefficient (F IS ) estimate at 0.31, suggesting that it is highly inbred.
In contrast, the other seven populations had allelic richness (A) ranging from 2.11 to 2.52, observed heterozygosity ranging from 0.51 to 0.64, and inbreeding coefficient (F IS ) ranging from 0.04 to 0.13, indicating that they are predominantly outbred.
Genetic differentiation
The genetic relationships among the 48 individuals showed a strong phylogeographical signal. Individuals from distinct populations generally clustered together in the neighbor joining tree (Fig. 2). An exception was found in the individuals from Poris de Abona (pop E) and Güimar (pop F). Most of the individuals from these two populations grouped together, likely due to high gene flow between the two populations. Another interesting pattern is the grouping of two individuals from Poris de Abona (pop E) with two individuals from Punta de Arenas Blancas (pop B), possibly indicative of a gene exchange event in the past. The former is located in Tenerife and the latter in the island of El Hierro.

Neighbor joining (NJ) tree showing the relationships of all 48 L. sessilifolius samples based on the 11 microsatellite loci. The genetic distances were calculated using the Lynch distance method (Lynch 1990). Key A = Mazo (La Palma), B = Punta de Arenas Blancas (El Hierro), C = Tejina-Milán (Tenerife), D = Puntallana (La Gomera), E = Poris de Abona (Tenerife), F = Güimar (Tenerife), G = Tecorone (El Hierro), H = Barranco de Ruiz (Tenerife)
Pairwise genetic distance measures between the eight populations based on F ST and other metrics showed that populations from the same islands clustered together (Figs. 3, 4). In the UPGMA and neighbour joining trees the populations from each island generally group together, with the exception of population Tejina-Milán (C), which is located on the Anaga peninsula in Tenerife and is genetically distinct from all the other L. sessilifolius populations.

Dendrogram (UPGMA) based on F ST pairwise genetic distances calculated using the software POLYSAT. Key A = Mazo (La Palma), B = Punta de Arenas Blancas (El Hierro), C = Tejina-Milán (Tenerife), D = Puntallana (La Gomera), E = Poris de Abona (Tenerife), F = Güimar (Tenerife), G = Tecorone (El Hierro), H = Barranco de Ruiz (Tenerife)

Unrooted neighbour-joining trees showing the genetic relationships of the populations based on various distance measures. Key A = Mazo (La Palma), B = Punta de Arenas Blancas (El Hierro), C = Tejina-Milán (Tenerife), D = Puntallana (La Gomera), E = Poris de Abona (Tenerife), F = Güimar (Tenerife), G = Tecorone (El Hierro), H = Barranco de Ruiz (Tenerife)
Population genetic structure was analyzed using the Structure software (Fig. 5). The best inference of K (maximum ΔK) occurred at K = 7 with a value of 89.0, separating all the populations except the adjacent Tenerife populations E and F which appear as a single population. These two populations are located on the eastern mountainside of Tenerife (i.e. Güimar and Poris de Abona) and appear to have considerable gene flow between them. In comparison the next largest peak was K = 3 with a value of 8.7. At K = 3 the Tejina-Milán population (C) was grouped with the La Gomera population (D) and is thus separated from all the other Tenerife populations, which group together and with the La Palma population (A, E, F, H). The third group is formed by the El Hierro populations (B, G).

STRUCTURE plots at K = 3 and K = 7. Key A = Mazo (La Palma), B = Punta de Arenas Blancas (El Hierro), C = Tejina-Milán (Tenerife), D = Puntallana (La Gomera), E = Poris de Abona (Tenerife), F = Güimar (Tenerife), G = Tecorone (El Hierro), H = Barranco de Ruiz (Tenerife). (Color figure online)
Discussion
The island pattern
Of the islands in which L. sessilifolius is distributed, Tenerife is the oldest (11.6 Ma) followed by La Gomera (10 Ma), La Palma (2 Ma) and El Hierro (1 Ma) (Carracedo 1994; Juan et al. 2000; Fernández-Palacios et al. 2011). Figure 3 shows clearly that when multiple populations were examined for an island, they generally clustered together (El Hierro: G, B and Tenerife: E, F, H). The only exception is the divergent population from Anaga, Tenerife (Tejina-Milán, C in Fig. 3), which is discussed below. These nuclear microsatellites do not reflect the morphological division of L. sessilifolius DC. into two subspecies (L. sessilifolius subsp. villossisimus from El Hierro and subsp. sessilifolius from other islands) (see Fig. 1).
Tenerife was originally composed of three palaeoislands: Adeje (11.6–3.5 Ma), Teno (6.7–4.5 Ma), and Anaga (6.5–3.5 Ma) (Ancochea et al. 1990; Fernández-Palacios et al. 2011). These palaeoislands were then joined into the modern island of Tenerife by volcanic activity, a process that was complete by c. 2 Ma (Ancochea et al. 1990), with connection between the palaeoislands likely achieved by 3.5 Ma. The paleoislands of Tenerife, particularly Teno and Anaga, are very rich in local endemics (Trusty et al. 2005), feasibly due to the isolation of these lineages followed by endemic evolution prior to 3.5 Ma. Furthermore, a series of massive landslips (c. 170–800 ka) may have helped to keep the palaeoislands isolated, even after connection (Ancochea et al. 1990; Watts and Masson 1995; Juan et al. 2000).
It is therefore not surprising that the Anaga population (C: Tejina-Milán) of L. sessilifolius is highly distinctive. Cluster analysis and STRUCTURE are consistent in dividing L. sessilifolius populations in two: Tejina-Milán vs the rest. One hypothesis that would be consistent with our results is that L. sessilifolius evolved in Anaga and La Gomera and then spread to other islands and back-colonized Tenerife. In this context it should be mentioned that Anaga and Gomera formed part of a palaeo-archipelago in the past (Fernandez-Palacios et al. 2011). L. sessilifoliius has also been reported from Teno and Adeje but samples from these areas are not included here. Given the distinctiveness of the Anaga population, future investigation of the Teno and Adeje populations is warranted.
Gene flow between adjacent populations
It seems likely that considerable gene flow occurs between populations Poris de Abona (pop E) and Güimar (pop F). This is indicated by the intermixing of individuals from these two populations in the tree of Fig. 2, and the high diversity measures (including allelic richness) shown by each of these populations in Table 3. Gene flow between them is likely, as they are located near each other on the southeastern coast of Tenerife with no physical barriers separating them.
Tejina-Milán: a distinctive population with low genetic diversity in Tenerife
The Tejina-Milán population (Tenerife) is outstanding because of its high genetic differentiation with respect to the other Tenerife populations. In the UPGMA tree, it is sister to all other populations of L. sessilifolius. This distinctiveness of the Tejina-Milán population may indicate that this is an anciently diverged population. The origin of this distinct lineage may be explained by the geological history of the Anaga peninsula. The Anaga palaeoisland and La Gomera represent some of the oldest areas of the western Canary Islands (Carracedo and Day 2002). The topology of the UPGMA tree supports that the colonization sequence of the lineage of L. sessilifolius proceeded according to the ages of the geological substrates where it occurs, so that the other Tenerife populations would represent more recent colonization events, ultimately deriving from either La Gomera or Anaga. Previous dating of the diversification of the L. sessilifolius lineage (Ojeda et al. 2012) indicates that the group radiated within the last 2 Mya, around the time of the most recent volcanic activity in Tenerife.
The low genetic diversity of the Anaga population (population C) possibly indicates that this is a relictual population that has undergone a range contraction. Population C has a very restricted area and is isolated by surrounding populations of Lotus tenellus. Our findings are in contrast with the results in Canarina canariensis (L.) Vatke (Campanulaceae), which found higher genetic diversity on populations distributed on the paleo-islands of Anaga, Teno and Roque del Conde, suggesting that these populations likely represent ancestral lineages (Mairal et al. 2015). Within genus Lotus there are two other described species in the northeast side of Anaga (Lotus dumetorum and L. tenellus), but we have not observed hybrids between any of them and L. sessilifolius. In addition, with the exception of a single population of L. tenellus, we did not observe any populations of related species in the close vicinity of the Tejina-Milán population.
There are no obvious morphological distinctions between the Tejina-Milán and the other L. sessilifolius populations in Tenerife; however, if morphological characters were to be found distinguishing this population, then formal taxonomic recognition (for instance at subspecific level) should be considered. On the basis of the results presented here, especial conservation measures should be considered for this population. For instance it is important to maintain the distinctiveness of this population by avoiding the introduction of L. sessilifolius from other parts of the island to the Anaga peninsula Biosphere Reserve, and monitoring development so as not to allow the extinction of this population.
General implications for conservation genetics
Lotus sessilifolius is one of the most common and variable of the endemic Lotus species of Macaronesia. However, as noted above, the microsatellite variation has revealed deep genetic structuring which should be used to inform conservation. The Anaga peninsula has many locally endemic plant taxa, which are interpreted as resulting from geological history, such as Micromeria glomerata and M. rivas-martinezii (Puppo et al. 2014, 2016). However, in L. sessilifolius the variation is cryptic in that there are no obvious phenotypic differences. It is possible that there are further distinct populations of this lineage, and others, on the Anaga Peninsula yet to be discovered. Additional genetic analysis, including from the other palaeoislands, Teno and Adeje, may therefore uncover more cryptic diversity in these regions.
In addition, the microsatellite primers described here are potentially of great value for conservation genetic studies of the many rare and endangered Lotus species of Macaronesia (Table 4). Initial tests suggest that the primers amplify successfully in all the species listed in Table 4. Many Canarian Lotus species have narrow distributions and are often restricted to specific habitats within each island. Thus, the group is highly susceptible to habitat destruction, and at least 11 species are listed under some category of threat, from rare to critically endangered (Table 4) (VV. AA. 2000; Bañares et al. 2004; Martín et al. 2008). Despite the relatively high number of endangered species, only two previous studies have assessed the genetic diversity in six endemic species (three endangered) using allozymes (Oliva-Tejera et al. 2005, 2006). Of the six species analyzed, only the endangered L. kunkelii showed relatively low levels of genetic variability, presumably due to inbreeding, very low population size and geographic isolation (Oliva-Tejera et al. 2006). The remaining species analyzed showed high levels of genetic variability, comparable to those reported in other continental Lotus species (Gauthier et al. 1998). The microsatellite tools reported here will facilitate conservation genetics work on diverse Lotus species.
References
Allan GJ, Francisco-Ortega J, Santos-Guerra A, Boerner E, Zimmer EA (2004) Molecular phylogenetic evidence for the geographic origin and classification of Canary Island Lotus (Fabaceae: Loteae). Mol Phylogenet Evol 32:123–138
Ancochea E, Fuster J, Ibarrola E, Cendrero A, Coello J, Hernan F, Cantagrel JM, Jamond C (1990) Volcanic evolution of the island of Tenerife (Canary Islands) in the light of new K-Ar data. J Volcanol Geotherm Res 44:231–249
Ashley MV (2010) Plant parentage, pollination, and dispersal: how DNA microsatellites have altered the landscape. Crit Rev Plant Sci 29:148–161
Bañares A, Blanca G, Güemes J, Moreno JC, Ortiz S (2004) Atlas y Libro Rojo De La Flora Vascular Amenazada de España. Dirección General de Conservación de la Naturaleza, Madrid
Bruvo R, Michiels NK, D’Souza TG, Schulenburg H (2004) A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol Ecol 13:2101–2106
Carracedo JC (1994) The Canary Islands: an example of structural control on the growth of large oceanic island volcanos. J Volcanol Geotherm Res 60:225–241
Carracedo JC, Day S (2002) Canary Islands. Terra Publishing, Harpenden
Clark LV, Jasieniuk M (2011) POLYSAT: an R package for polyploid microsatellite analysis. Mol Ecol Resour 11:562–566
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem Bull 19:11–15
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fernández-Palacios JM, de Nascimento L, Otto R, Delgado JD, García-del-Rey E, Arévalo JR, Whittaker RJ (2011) A reconstruction of Palaeo-Macaronesia, with particular reference to the long-term biogeography of the Atlantic island laurel forests. J Biogeogr 38:226–246
García-Verdugo C, Sajeva M, La Mantia T, Harrouni C, Msanda F, Caujapé-Castells J (2015) Do island plant populations really have lower genetic variation than mainland populations? Effects of selection and distribution range on genetic diversity estimates. Mol Ecol 24:726–741
Gauthier P, Lumaret R, Bedecarrats A (1998) Genetic variation and gene flow in Alpine diploid and tetraploid populations of Lotus (L. alpinus (DC) Schleicher, L. corniculatus L). 1. Insights from morphological and allozyme markers. Heredity 80:683–693
Goldstein DB, Linares AR, Cavalli-Sforza LL, Feldman MW (1995) An evaluation of genetic distances for use with microsatellite loci. Genetics 139:463–471
Huelsenbeck JP, Kirkpatrick M (1996) Do phylogenetic methods produce trees with biased shapes? Evol Int J Org Evol 50:1418–1424
Juan C, Emerson BC, Oromí P, Hewitt GM (2000) Colonization and diversification: towards a phylogeographic synthesis for the Canary Islands. Trends Ecol Evol 15:104–109
Kramer AT, Fant JB, Ashley MV (2011) Infliuences of landscape and pollinators on population genetic structure: examples from three Penstemon (Plantaginaceae) species in the Great Basin. Am J Bot 98:109–121
Lynch M (1990) The similarity index and DNA fingerprinting. Mol Biol Evol 7:478–484
Mairal M, Sanmartin I, Aldasoro JJ, Culshaw V, Manolopoulou I, Alarcon M (2015) Palaeo-islands as refugia and sources of genetic diversity within volcanic archipelagos: the case of the widespread endemic Canarina canariensis (Campanulaceae). Mol Ecol 24:3944–3963
Martín JL, Arechavaleta M, Borges PA, Faria B (2008) Top 100. Las 100 especies amenazadas prioritarias de gestión en la región europea biogeográfica de la Macaronesia. Consejería de Medio Ambiente y Ordenación Territorial, Gobierno de Canarias, Gran Canaria
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Ochieng JW, Steane DA, Ladiges PY, Baverstock PR, Henry RJ, Shepherd M (2007) Microsatellites retain phylogenetic signals across genera in eucalypts (Myrtaceae). Gene Mol Biol 30:1125–1134
Ojeda I, Santos-Guerra A, Jaen-Molina R, Oliva-Tejera F, Caujape-Castells J, Cronk Q (2012) The origin of bird pollination in Macaronesian Lotus (Loteae, Leguminosae). Mol Phylogenet Evol 62:306–318
Oliva-Tejera F, Caujape-Castells J, Naranjo-Suarez J, Navarro-Deniz J, Acebes-Ginoves JR, Bramwell D (2005) Population genetic differentiation in taxa of Lotus (Fabaceae: Loteae) endemic to the Gran Canarian pine forest. Heredity 94:199–206
Oliva-Tejera F, Caujape-Castells J, Navarro-Deniz J, Reyes-Betancort A, Scholz S, Baccarani-Rosas M, Cabrera-Garcia N (2006) Patterns of genetic divergence of three Canarian endemic Lotus (Fabaceae): implications for the conservation of the endangered L. kunkelii. Am J Bot 93:1116–1124
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290
Petren K, Grant BR, Grant PR (1999) A phylogeny of Darwin’s finches based on microsatellite DNA length variation. Proc R Soc (Lond) B 266:321–329
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puppo P, Curto M, Velo-Anton G, de Paz PLP, Meimberg H (2014) The influence of geological history on diversification in insular species: genetic and morphological patterns of Micromeria Benth. (Lamiaceae) in Tenerife (Canary archipelago). J Biogeogr 41:1871–1882
Puppo P, Curto M, Meimberg H (2016) Genetic structure of Micromeria (Lamiaceae) in Tenerife, the imprint of geological history and hybridization on within-island diversification. Ecol Evol 6:3443–3460
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Sandral G, Remizova MV, Sokoloff DD (2006) A taxonomic survey of Lotus section Pedrosia (Leguminose, Loteae). Wulfenia 13:97–192
Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, Nakao M, Sasamoto S, Watanabe A, Ono A, Kawashima K, Fujishiro T, Katoh M, Kohara M, Kishida Y, Minami C, Nakayama S, Nakazaki N, Shimizu Y, Shinpo S, Takahashi C, Wada T, Yamada M, Ohmido N, Hayashi M, Fukui K, Baba T, Nakamichi T, Mori H, Tabata S (2008) Genome structure of the legume, Lotus japonicus. DNA Res 15:227–239
Sosa PA, Gonzalez-Perez MA, Moreno C, Clarke JB (2010) Conservation genetics of the endangered endemic Sambucus palmensis Link (Sambucaceae) from the Canary Islands. Conserv Genet 11:2357–2368
Thrall PH, Young A (2000) AUTOTET: A program for analysis of autotetraploid genotypic data. J Hered 91:348–349
Trusty JL, Olmstead RG, Santos-Guerra AR, Sá-Fontinha S, Francisco-Ortega JA (2005) Molecular phylogenetics of the Macaronesian-endemic genus Bystropogon (Lamiaceae): palaeo-islands, ecological shifts and interisland colonizations. Mol Ecol 14:1177–1189
VV. AA. (2000) Lista Roja de Flora Vascular Española (valoración según categorías UICN). Conservación Vegetal 6 (extra):11–38
Watts AB, Masson DG (1995) A giant landslide on the north flank of Tenerife, Canary Islands. J Geophys Res 100:24487–24498
Acknowledgements
This work was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC) under the Discovery Grants Program (Grant No. RGPIN-2014-05820 to QC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yang, J.Y., Ojeda, D.I., Santos-Guerra, A. et al. Population differentiation in relation to conservation: nuclear microsatellite variation in the Canary Island endemic Lotus sessilifolius (Fabaceae). Conservation Genet Resour 10, 219–227 (2018). https://doi.org/10.1007/s12686-017-0778-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-017-0778-1