
Abstract
Nibrin (NBN), located on chromosome 8q21 is a gene involved in DNA double-strand break repair that has been implicated in the rare autosomal recessive chromosomal instability syndrome known as Nijmegen Breakage Syndrome (NBS). NBS is characterized by specific physical characteristics (microcephaly and dysmorphic facies), immunodeficiency, and increased risk of malignancy. Individuals who are heterozygous for NBN mutations are clinically asymptomatic, but may display an elevated risk for certain cancers including, but not limited to, ovarian and prostate cancer as well as various lymphoid malignancies. In this study, 94 unrelated familial prostate cancer cases from the University of Michigan Prostate Cancer Genetics Project (n = 54) and Johns Hopkins University (n = 40) were subjected to targeted next-generation sequencing of the exons, including UTRs, of NBN. One individual of European descent, diagnosed with prostate cancer at age 52, was identified to have a heterozygous 2117 C > G mutation in exon 14 of the gene, that results in a premature stop at codon 706 (S706X). Sequencing of germline DNA from additional male relatives showed partial co-segregation of the NBN S706X mutation with prostate cancer. This NBN mutation was not observed among 2768 unrelated European men (1859 with prostate cancer and 909 controls). NBN is involved in double-strand break repair as a component of the MRE11 (meiotic recombination 11)/RAD50/NBN genomic stability complex. The S706X mutation truncates the protein in a highly conserved region of NBN near the MRE11 binding site, thus suggesting a role for rare NBN mutations in prostate cancer susceptibility.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prostate cancer is the most common non-cutaneous cancer diagnosed among American men and the second leading cause of cancer death with an estimated 241,740 new cases and 28,170 deaths expected in the United States in 2012 [1]. In addition to increasing age and African American race, family history is also a recognized risk factor for prostate cancer. The diagnosis of sporadic prostate cancer cases remains high in the general population, suggesting that disease risk may be influenced by additional risk factors such as diet, obesity, and environment. However, familial aggregation of prostate cancer suggests that in some cases, heritable genetic factors may play a key role in the development of prostate cancer.
Initial segregation analyses of familial prostate cancer concluded that inherited predisposition was due to the presence of at least one highly penetrant autosomal dominant prostate cancer predisposition gene [2–4]. Though subsequent linkage (e.g. HPC1 [5], HPCX [6], HPC20 [7]) and association (e.g. [8, 9, 10]) studies have revealed several prostate cancer susceptibility loci, these results have not been strongly replicated nor have they led to the identification of a causative gene mutation(s) capable of explaining a large percentage of familial risk. Evidence from these and other studies suggest that familial prostate cancer is a complex heterogeneous disease that may be influenced by the inheritance of less common risk loci with moderate penetrance. Next-generation sequencing of candidate gene regions can be useful in identifying rare variants that may explain prostate cancer risk in a small number of families.
Nijmegen breakage syndrome (NBS) is a rare autosomal recessive disorder that is characterized by microcephaly, immunodeficiency, chromosomal instability and sensitivity to ionizing radiation [11]. The gene for Nijmegen breakage syndrome (NBS1), also known as Nibrin (NBN) is located on chromosome 8q21; mutations in NBN are known to cause NBS [12]. To date, several NBS-causing mutations have been identified, the most prevalent being the NBN founder mutation 657del5 which is estimated to account for more than 90 % of all mutant alleles [13]. This mutation, found in exon 6 of NBN, is most often observed in members of the Slavic population which includes people from Poland, Ukraine, and the Czech Republic. Individuals heterozygous for NBN mutations are clinically asymptomatic however; there have been reports of increased incidence of various malignancies [14], particularly breast, prostate, colorectal, and lymphoid cancers (especially lymphoblastic leukemia and non-Hodgkin’s lymphoma). In the present study, we discuss the identification of a novel truncating mutation in NBN as a result of targeted next-generation sequencing of 94 familial prostate cancer cases from the University of Michigan and the Johns Hopkins University.
Materials and methods
Patient selection
University of Michigan prostate cancer genetics project (UM-PCGP)
UM-PCGP prostate cancer cases were restricted to (1) men diagnosed with prostate cancer with at least one living first- or second-degree relative also diagnosed with prostate cancer or (2) men diagnosed with prostate cancer at <56 years of age irrespective of family history. The diagnosis of prostate cancer was confirmed by medical record review whenever possible. All subjects provided written informed consent to participate in the study and all protocol and consent documents were approved by the University of Michigan Medical School Institutional Review Board.
Johns Hopkins University (JHU)
Hereditary prostate cancer (HPC) families each had at least three first-degree relatives affected with prostate cancer. The diagnosis of prostate cancer was verified by medical record review. Radical prostatectomy cases were men who underwent surgery for treatment of prostate cancer at the Johns Hopkins Hospital (JHH). Controls for this study consisted of men who underwent screening for prostate cancer, including measuring serum prostate-specific antigen (PSA) levels and digital rectal examination at JHH, Johns Hopkins Bayview Medical Center, Johns Hopkins University Applied Physics Laboratory (Columbia, MD) and several other locations in the mid-Atlantic area. Additional inclusion criteria required control subjects to have knowledge of prostate cancer ancestry by self-report and lack a prostate cancer diagnosis. For all JHU studies, research proposals were reviewed and approved by the institutional review board.
Targeted sequencing of NBN gene
We selected the youngest prostate cancer case with available DNA from 94 prostate cancer families (40 families from JHU and 54 from the UM-PCGP) [15]. Seven families were of African descent, 2 were of Asian descent, and the remaining 85 were of European descent. A primer library was designed for amplification of 8068 base pairs (bp) of NBN including all coding regions, exons, intron/exon boundaries, and 5′ and 3′ untranslated regions. We used the RainDance RDT 1000 system (RainDance Technologies, Inc., Lexington, MA) to amplify 3 μg of sheared genomic DNA from each sample using our primer library. Purified amplicons were used as templates for sequencing using the Life Technologies SOLiD™ system version 4.0 fragment library methodologies (Life Technologies Corporation, Carlsbad, CA). Sequence data processing was performed using Bioscope to align the sequences to the genomic reference (Build 36, hg18). Variant detection was performed using SamTools 1.3 [16] and SolSNP 1.1. We confirmed and tested all variant sequences in family members using standard Sanger sequencing, capillary electrophoresis technology and BigDye® Terminator chemistry (Applied Biosystems, Carlsbad, CA).
Genotyping of NBN variants
We genotyped identified NBN variants using the MassARRAY system (Sequenom, San Diego CA).
Results
Analysis of NBN revealed a novel heterozygous 2117 C > G mutation (Fig. 1) in a man diagnosed with prostate cancer at age 52 (NM_002485). The nonsense mutation is a single C to G transversion in exon 14 resulting in a coding change from TCA to TGA (Serine 706 Stop or S706X). This mutation, which codes for a truncated NBN protein that lacks the C-terminal ataxia-telangiectasia mutated (ATM) recruitment motif, was not present in 93 additional HPC probands.

Sequence chromatogram of exon 14 of NBN in the proband demonstrating the 2117 C > G mutation (a) and wild-type sequence (b). The arrow indicates the location of the base substitution
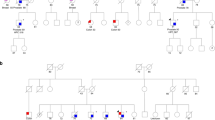
The pedigree of the family in which the NBN mutation was identified is depicted in Fig. 2, and features four individuals diagnosed with prostate cancer, as well as one case each of lymphoma, bladder cancer, and melanoma. Subsequent Sanger sequencing of DNA samples collected from three additional family members revealed that the proband’s father, who had been diagnosed with prostate and bladder cancer, and a brother who remains unaffected at age 59, were both carriers of the S706X mutation; however the proband’s paternal uncle with prostate cancer, diagnosed at age 70, was not a carrier (Fig. 2). Neither of the affected men who were NBN S706X carriers had intermediate or high risk prostate cancer based on available clinical information. DNA samples from the proband’s paternal aunt with non-Hodgkin’s lymphoma and son with melanoma were not available for testing.

Pedigree segregating the NBN S706X mutation. The proband initially selected for sequencing is indicated by the arrow. With the exception of the proband, the ages of diagnosis have been rounded to the nearest 5-year interval and are shown under the subject. The remaining symbols are described in the key. NHL non-Hodgkins lymphoma
The NBN S706X mutation was not observed among 1859 men with prostate cancer (JHU radical prostatectomy cases) and 909 male controls, all of whom describe themselves to be of European descent. Coding variants that were detected as a result of targeted next-generation sequencing of NBN in 85 individuals of European descent are summarized in Table 1.
Discussion
Targeted next-generation sequencing of the NBN gene resulted in the identification of a novel nonsense mutation S706X in one of 94 HPC families. The mutation was identified in two of three family members diagnosed with prostate cancer and therefore demonstrated incomplete segregation with prostate cancer in this pedigree.
A previous study by Cybulski et al. [17] implicated NBN as a prostate cancer susceptibility gene due to evidence of increased prostate cancer incidence in homozygous carriers of the 657del5 Eastern European founder mutation. The NBN 657del5 mutation was evaluated in Polish individuals and was found in 9 of 340 unselected patients with prostate cancer (2.6 %) compared with only 9 of 1500 (0.6 %) control subjects from the general population (OR = 4.5; 95 % confidence interval (1.7–11.5); p = 0.002) A follow-up study by Hebbring et al. [18] could not confirm this hypothesis due to the inability to identify the 657del5 mutation in DNA samples from 293 unaffected members of prostate cancer families and 697 control samples.
Nibrin or NBN is a component of the hMRE11 (meiotic recombination 11)/hRad50/NBN protein complex that is involved in initiating a response to DNA damage and is linked to DNA double-strand break repair [19]. Full length NBN consists of 754 amino acids and has a molecular weight of 95 kDa. The protein has three regions: the N-terminus, the central region, and the C-terminus (Fig. 3a). The N-terminal domain contains a fork-head associated (FHA) domain (amino acids 24–109) and two breast cancer C-terminus (BRCT) domains (amino acids 114–183 and 221–291) [20–22]. The central region is composed of consensus sequences for phosphorylation by ataxia-telangiectasia mutated (ATM) or ataxia-telangiectasia and Rad3-related (ATR) kinases [23]. The C-terminal region contains an MRE11 (amino acids 665–693) binding domain [24] and an ATM (amino acids 734–754) recruitment motif [25].

The NBN S706X mutation results in deletion of the ATM recruitment motif. a The N-terminus of the NBN protein contains a fork-head associated (FHA) domain and two breast cancer C-terminus (BRCT) domains. The C-terminal region contains an MRE11 binding domain (MBD) and an ATM recruitment motif (ARM). b Novel nonsense mutation S706X affects an amino acid located in the C-terminus in a highly conserved region of NBN
The S706X mutation results in a truncated NBN protein that lacks the extreme C-terminal ATM recruitment motif (ARM). The ATM protein kinase plays a role in suppressing chromosomal abnormalities by functioning as an agent of DNA damage response [26]. Whereas ATR is activated by single-strand breaks, ATM appears to be specific to double strand breaks (DSBs). The hMRE11/hRad50/NBN protein complex requires the highly conserved C-terminal 20 amino acids of NBN (Fig. 3b), which comprise the ARM, for interaction with ATM [25]. In the absence of the full-length protein, ATM is not actively recruited to the site of DNA damage, thus preventing efficient DSB repair. In addition, though heterozygous carriers of NBN mutations maintain the ability to produce full length NBN, the resulting truncation may be sufficient to promote tumorigenesis by means of haplo-insufficiency. Note that in the original report by Cybulski et al. [17], the majority (seven of eight) of the prostate cancer tumors from men who were heterozygous carriers of the founder NBN 657del5 mutation demonstrated loss of heterozygosity suggesting that NBN is acting as a classical tumor suppressor gene to promote cancer growth. Future tumor as well as in vitro studies and animal models should address this issue [27].
In the present study, we focused on identifying germline variants in NBN, an established prostate cancer candidate gene. Heterozygous carriers of deleterious NBN mutations have been shown to be at increased risk for several common malignancies including prostate cancer. Based on its deletion of the ATM recruitment motif, the S706X mutation is expected to impair the function of the highly conserved protein complex necessary for DNA damage response and subsequent DSB repair. Mutations in genes involved in DSB repair such as BRCA1, BRCA2 [28], and BRIP1 [29] have also been shown to increase risk of prostate cancer in some pedigrees. Exome sequencing of multiplex prostate cancer pedigrees may uncover additional deleterious variants in DNA repair genes that may contribute to this common disease. Although these variants are expected to be rare, knowledge of these mutations may be useful for risk assessment and may provide unique insights into potential therapeutic strategies [30].
References
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62(1):10–29. doi:10.3322/caac.20138
Carter BS, Beaty TH, Steinberg GD, Childs B, Walsh PC (1992) Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci USA 89(8):3367–3371
Schaid DJ, McDonnell SK, Blute ML, Thibodeau SN (1998) Evidence for autosomal dominant inheritance of prostate cancer. Am J Hum Genet 62(6):1425–1438. doi:10.1086/301862
Gronberg H, Damber L, Damber JE, Iselius L (1997) Segregation analysis of prostate cancer in Sweden: support for dominant inheritance. Am J Epidemiol 146(7):552–557
Smith JR, Freije D, Carpten JD, Gronberg H, Xu J, Isaacs SD, Brownstein MJ, Bova GS, Guo H, Bujnovszky P, Nusskern DR, Damber JE, Bergh A, Emanuelsson M, Kallioniemi OP, Walker-Daniels J, Bailey-Wilson JE, Beaty TH, Meyers DA, Walsh PC, Collins FS, Trent JM, Isaacs WB (1996) Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science 274(5291):1371–1374
Xu J, Meyers D, Freije D, Isaacs S, Wiley K, Nusskern D, Ewing C, Wilkens E, Bujnovszky P, Bova GS, Walsh P, Isaacs W, Schleutker J, Matikainen M, Tammela T, Visakorpi T, Kallioniemi OP, Berry R, Schaid D, French A, McDonnell S, Schroeder J, Blute M, Thibodeau S, Gronberg H, Emanuelsson M, Damber JE, Bergh A, Jonsson BA, Smith J, Bailey-Wilson J, Carpten J, Stephan D, Gillanders E, Amundson I, Kainu T, Freas-Lutz D, Baffoe-Bonnie A, Van Aucken A, Sood R, Collins F, Brownstein M, Trent J (1998) Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet 20(2):175–179. doi:10.1038/2477
Berry R, Schroeder JJ, French AJ, McDonnell SK, Peterson BJ, Cunningham JM, Thibodeau SN, Schaid DJ (2000) Evidence for a prostate cancer-susceptibility locus on chromosome 20. Am J Hum Genet 67(1):82–91. doi:10.1086/302994
Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hayes RB, Hunter DJ, Chanock SJ (2008) Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 40(3):310–315. doi:10.1038/ng.91
Eeles RA, Kote-Jarai Z, Al Olama AA, Giles GG, Guy M, Severi G, Muir K, Hopper JL, Henderson BE, Haiman CA, Schleutker J, Hamdy FC, Neal DE, Donovan JL, Stanford JL, Ostrander EA, Ingles SA, John EM, Thibodeau SN, Schaid D, Park JY, Spurdle A, Clements J, Dickinson JL, Maier C, Vogel W, Dork T, Rebbeck TR, Cooney KA, Cannon-Albright L, Chappuis PO, Hutter P, Zeegers M, Kaneva R, Zhang HW, Lu YJ, Foulkes WD, English DR, Leongamornlert DA, Tymrakiewicz M, Morrison J, Ardern-Jones AT, Hall AL, O’Brien LT, Wilkinson RA, Saunders EJ, Page EC, Sawyer EJ, Edwards SM, Dearnaley DP, Horwich A, Huddart RA, Khoo VS, Parker CC, Van As N, Woodhouse CJ, Thompson A, Christmas T, Ogden C, Cooper CS, Southey MC, Lophatananon A, Liu JF, Kolonel LN, Le Marchand L, Wahlfors T, Tammela TL, Auvinen A, Lewis SJ, Cox A, FitzGerald LM, Koopmeiners JS, Karyadi DM, Kwon EM, Stern MC, Corral R, Joshi AD, Shahabi A, McDonnell SK, Sellers TA, Pow-Sang J, Chambers S, Aitken J, Gardiner RA, Batra J, Kedda MA, Lose F, Polanowski A, Patterson B, Serth J, Meyer A, Luedeke M, Stefflova K, Ray AM, Lange EM, Farnham J, Khan H, Slavov C, Mitkova A, Cao G, Easton DF (2009) Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet 41(10):1116–1121. doi:10.1038/ng.450
Kote-Jarai Z, Olama AA, Giles GG, Severi G, Schleutker J, Weischer M, Campa D, Riboli E, Key T, Gronberg H, Hunter DJ, Kraft P, Thun MJ, Ingles S, Chanock S, Albanes D, Hayes RB, Neal DE, Hamdy FC, Donovan JL, Pharoah P, Schumacher F, Henderson BE, Stanford JL, Ostrander EA, Sorensen KD, Dork T, Andriole G, Dickinson JL, Cybulski C, Lubinski J, Spurdle A, Clements JA, Chambers S, Aitken J, Gardiner RA, Thibodeau SN, Schaid D, John EM, Maier C, Vogel W, Cooney KA, Park JY, Cannon-Albright L, Brenner H, Habuchi T, Zhang HW, Lu YJ, Kaneva R, Muir K, Benlloch S, Leongamornlert DA, Saunders EJ, Tymrakiewicz M, Mahmud N, Guy M, O’Brien LT, Wilkinson RA, Hall AL, Sawyer EJ, Dadaev T, Morrison J, Dearnaley DP, Horwich A, Huddart RA, Khoo VS, Parker CC, Van As N, Woodhouse CJ, Thompson A, Christmas T, Ogden C, Cooper CS, Lophatonanon A, Southey MC, Hopper JL, English DR, Wahlfors T, Tammela TL, Klarskov P, Nordestgaard BG, Roder MA, Tybjaerg-Hansen A, Bojesen SE, Travis R, Canzian F, Kaaks R, Wiklund F, Aly M, Lindstrom S, Diver WR, Gapstur S, Stern MC, Corral R, Virtamo J, Cox A, Haiman CA, Le Marchand L, Fitzgerald L, Kolb S, Kwon EM, Karyadi DM, Orntoft TF, Borre M, Meyer A, Serth J, Yeager M, Berndt SI, Marthick JR, Patterson B, Wokolorczyk D, Batra J, Lose F, McDonnell SK, Joshi AD, Shahabi A, Rinckleb AE, Ray A, Sellers TA, Lin HY, Stephenson RA, Farnham J, Muller H, Rothenbacher D, Tsuchiya N, Narita S, Cao GW, Slavov C, Mitev V, Easton DF, Eeles RA (2011) Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat Genet 43(8):785–791. doi:10.1038/ng.882ng.882
Weemaes CM, Hustinx TW, Scheres JM, van Munster PJ, Bakkeren JA, Taalman RD (1981) A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr Scand 70(4):557–564
Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanova E, Cooper PR, Nowak NJ, Stumm M, Weemaes CM, Gatti RA, Wilson RK, Digweed M, Rosenthal A, Sperling K, Concannon P, Reis A (1998) Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93(3):467–476. doi:S0092-8674(00)81174-5
Antoccia A, Kobayashi J, Tauchi H, Matsuura S, Komatsu K (2006) Nijmegen breakage syndrome and functions of the responsible protein, NBS1. Genome Dyn 1:191–205. doi:10.1159/000092508
Seemanova E (1990) An increased risk for malignant neoplasms in heterozygotes for a syndrome of microcephaly, normal intelligence, growth retardation, remarkable facies, immunodeficiency and chromosomal instability. Mutat Res 238(3):321–324
Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, Wang Y, Bizon C, Yan G, Gielzak M, Partin AW, Shanmugam V, Izatt T, Sinari S, Craig DW, Zheng SL, Walsh PC, Montie JE, Xu J, Carpten JD, Isaacs WB, Cooney KA (2012) Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med 366(2):141–149. doi:10.1056/NEJMoa1110000
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. doi:10.1093/bioinformatics/btp352
Cybulski C, Gorski B, Debniak T, Gliniewicz B, Mierzejewski M, Masojc B, Jakubowska A, Matyjasik J, Zlowocka E, Sikorski A, Narod SA, Lubinski J (2004) NBS1 is a prostate cancer susceptibility gene. Cancer Res 64(4):1215–1219
Hebbring SJ, Fredriksson H, White KA, Maier C, Ewing C, McDonnell SK, Jacobsen SJ, Cerhan J, Schaid DJ, Ikonen T, Autio V, Tammela TL, Herkommer K, Paiss T, Vogel W, Gielzak M, Sauvageot J, Schleutker J, Cooney KA, Isaacs W, Thibodeau SN (2006) Role of the Nijmegen breakage syndrome 1 gene in familial and sporadic prostate cancer. Cancer Epidemiol Biomarkers Prev 15(5):935–938. doi:10.1158/1055-9965.EPI-05-0910
Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR 3rd, Hays L, Morgan WF, Petrini JH (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93(3):477–486. doi:S0092-8674(00)81175-7
Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV (1997) A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. Faseb J 11(1):68–76
Callebaut I, Mornon JP (1997) From BRCA1 to RAP1: a widespread BRCT module closely associated with DNA repair. FEBS Lett 400(1):25–30. doi:S0014-5793(96)01312-9
Becker E, Meyer V, Madaoui H, Guerois R (2006) Detection of a tandem BRCT in Nbs1 and Xrs2 with functional implications in the DNA damage response. Bioinformatics 22(11):1289–1292. doi:10.1093/bioinformatics/btl075
Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB (2000) ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 404(6778):613–617. doi:10.1038/35007091
Desai-Mehta A, Cerosaletti KM, Concannon P (2001) Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol Cell Biol 21(6):2184–2191. doi:10.1128/MCB.21.6.2184-2191.2001
Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434(7033):605–611. doi:10.1038/nature03442
Bennardo N, Stark JM (2010) ATM limits incorrect end utilization during non-homologous end joining of multiple chromosome breaks. PLoS Genet 6(11):e1001194. doi:10.1371/journal.pgen.1001194
Berger AH, Pandolfi PP (2011) Haplo-insufficiency: a driving force in cancer. J Pathol 223(2):137–146. doi:10.1002/path.2800
Liede A, Karlan BY, Narod SA (2004) Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: a review of the literature. J Clin Oncol 22(4):735–742. doi:10.1200/JCO.2004.05.055JCO.2004.05.055
Kote-Jarai Z, Jugurnauth S, Mulholland S, Leongamornlert DA, Guy M, Edwards S, Tymrakiewitcz M, O’Brien L, Hall A, Wilkinson R, Al Olama AA, Morrison J, Muir K, Neal D, Donovan J, Hamdy F, Easton DF, Eeles R (2009) A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer. Br J Cancer 100(2):426–430. doi:10.1038/sj.bjc.6604847
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS (2009) Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361(2):123–134. doi:10.1056/NEJMoa0900212
Acknowledgments
This work was supported by grants (CA79596, CA136621, and P50 CA69568) from the National Institutes of Health. The authors would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). We also thank the men with prostate cancer and their family members for their participation in this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zuhlke, K.A., Johnson, A.M., Okoth, L.A. et al. Identification of a novel NBN truncating mutation in a family with hereditary prostate cancer. Familial Cancer 11, 595–600 (2012). https://doi.org/10.1007/s10689-012-9555-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-012-9555-1