
Abstract
Fusarium head blight (FHB) is one of the most serious diseases in small-grain cereals including triticale (× Triticosecale Wittmack). The disease reduces yield and accumulates mycotoxins which are harmful to human and animal health. Triticale grain is almost exclusively used on-farm in feed formulations for swine and other animals, and swine is the most susceptible farm animal to Fusarium mycotoxins. In order to evaluate the potential of genomics-assisted breeding to FHB, we performed the first genome-wide association study for FHB resistance in triticale. QTL for FHB resistance were identified on chromosomes 2A, 2B, 5B and 3R with an explained genotypic variance ranging from 0.28 to 30.23% and a total explained genetic variance of 56.64%. A QTL on chromosome 3R that explained 15.38% of the genotypic variance was identified for the first time. Association mapping was complemented by genome-wide prediction, which yielded a high prediction accuracy of 0.78 for FHB resistance when weighted genomic selection was performed. Collectively our findings highlight the potential of genomics-assisted approaches to improve Fusarium resistance in triticale in early generations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Triticale (× Triticosecale Wittmack) is an amphiploid man-made hybrid between wheat (Triticum spp.) as seed-parent and rye (Secale cereale L.) as pollen-donor parent. The initial idea of this intergeneric cross was to combine the high robustness of rye with the great yield potential and excellent baking quality of wheat (Mergoum et al. 2009). Nowadays, however, most commercially grown triticale originate from crosses between tetraploid wheat (Triticum turgidum L. var. durum, AABB) and diploid rye (Secale cereale, RR), resulting in hexaploid triticale (AABBRR) (Oettler 2005). The lack of D-genome chromosomes, which largely determine baking quality in Triticum aestivum, explains why triticale has never become a serious substitute of wheat as a food grain (Oettler 2005). Nevertheless, triticale is a highly valuable source of livestock feed due to its high starch digestibility and feed conversion efficiency (Bird et al. 1999; Oettler 2005; Mergoum et al. 2009). In particular, triticale grain is mainly used on-farm as an excellent component in swine feed formulations. Moreover, triticale has shown great potential for biomass production (Gowda et al. 2011; Pronyk and Mazza 2011). The economic importance of triticale is reflected by a global acreage of around 4.4 million hectares and a production of 15.7 million tons in 2016 (FAOSTAT 2018). Europe is the main triticale producer and accounts for 90% of the world production (Miedaner et al. 2016; FAOSTAT 2018), where Poland is by far the biggest producer, followed by Germany and Belarus (FAOSTAT 2018).
Triticale has always been a susceptible host to diseases caused by necrotrophic pathogens, such as Septoria tritici blotch, Stagonospora nodorum blotch and Fusarium head blight (FHB) (Kalih et al. 2015). The latter, caused by Fusarium spp., draws special attention due to the reduction of yield components and production of a broad range of mycotoxins that are harmful to animals and humans (Oettler and Wahle 2001; Audenaert et al. 2013; Miedaner et al. 2016). Among the Fusarium species, Fusarium graminearum is the most important, followed in Europe by F. culmorum and F. poae (Becher et al. 2013). Fusarium graminearum and F. culmorum produce deoxynivalenol (DON) or nivalenol (NIV), zearalenone (ZON), and other mycotoxins (Scherm et al. 2013; Miedaner et al. 2013).
Among farm animals, swine is the most susceptible to Fusarium mycotoxins. Prolonged diets with FHB-contaminated triticale can result in complications of the immune and nervous system, female infertility and fetus malformation (Čonková et al. 2003; Bennett and Klich 2003; EFSA 2004; Sugita-Konishi et al. 2008; Becher et al. 2013). Due to the negative impacts on human and animal health, the European Union established an orientation value of maximum 0.9 mg DON kg−1 cereal for swine, which is even lower than the one stipulated for human nutrition, which is 1.25 mg DON kg−1 (European Commission 2006; Miedaner et al. 2016). Fungicide application is not fully effective to mitigate FHB and can be costly to the farmer and to the environment (Becher et al. 2013). Thus, varietal resistance is the most promising option to control disease spread and mycotoxin accumulation. However, it must be integrated with other disease management techniques such as crop rotation and soil tillage in order to be fully efficient (Bolduan et al. 2009; Miedaner et al. 2016). The negative impact on swine production in combination with the lack of available highly effective fungicides leads to the urgency of identifying triticale cultivars resistant to FHB.
Previous studies confirmed the presence of sufficient genotypic variation for Fusarium resistance in triticale, with a moderate to high heritability under artificial infection (Oettler and Wahle 2001; Kalih et al. 2014, 2015; Dhariwal et al. 2018). According to previous studies, FHB resistance is caused by the interaction of many genes with predominantly additive gene action (Oettler et al. 2004). This suggests recurrent selection as a promising method to increase FHB resistance (Oettler and Wahle 2001). However, FHB resistance needs several scorings per season, shows covariation with flowering time (FT) (Boeven et al. 2016b; Miedaner et al. 2017) and plant height (PH), and a high genotype-by-environment interaction. Thus, robust phenotypic FHB data require several locations, replications, and artificial inoculations leading to high costs and a time-consuming disease assessment (Oettler and Wahle 2001; Miedaner et al. 2004; Martin et al. 2012).
Genomics-assisted breeding can be a promising option to increase selection efficiency for FHB resistance in triticale breeding programs. Many quantitative trait loci (QTL) studies have already been conducted for wheat and at least 160 QTL have been identified in more than 30 mapping populations (Löffler et al. 2009; Miedaner et al. 2016). By contrast, studies of quantitative resistance to FHB in triticale are far behind. To the best of our knowledge, only four linkage mapping studies for FHB resistance in triticale have been published to date (Kalih et al. 2014, 2015; Miedaner et al. 2016; Dhariwal et al. 2018). Besides linkage mapping, genome-wide association (GWA) mapping is a powerful method to identify QTL. It explores historical recombinations of the individuals leading to a higher mapping resolution and increased success rate of QTL transferability to other genetic backgrounds when compared to linkage mapping (Würschum 2012). Despite these potential advantages, no GWA study for FHB resistance in triticale has yet been published. Genomic selection (GS) is an alternative approach that is based on a simultaneous estimation of all marker effects, thereby capturing also the variance of small-effect QTL (Heffner et al. 2010). Considering that FHB resistance in triticale has a similar complex genetic architecture as in wheat (Mirdita et al. 2015), GS approaches have the potential to assist FHB breeding. However, knowledge about the potential of GS to assist FHB resistance breeding in triticale is lacking.
The aim of this study was to dissect the genetic architecture of FHB and to evaluate the potential of genomics-assisted approaches to improve resistance breeding programs in a triticale diversity panel. In particular, our objectives were to (1) analyze the effect of flowering time and plant height on FHB resistance, (2) dissect the genetic architecture of FHB resistance in triticale, (3) evaluate genomics-assisted approaches as tools to expand the selection portfolio, and (4) draw conclusions for current triticale breeding programs.
Materials and methods
Plant materials and field trials
This study comprised 133 diverse winter triticale cultivars and elite breeding lines. These lines are a subset of the 434 winter triticale lines and hybrids described in a previous study by Boeven et al. (2016b). Our subset of genotypes solely represents inbred lines originating from the following countries: Belarus, Canada, France, Germany, Netherlands, Poland, Romania, Russian Federation and Switzerland. Entries were sown in two-row observation plots with 1.2 m length and 0.41 m width between rows and with a sowing density of 30 kernels per genotype. Experiments were conducted in four environments as a combination of two locations and 2 years in the growing seasons 2013 and 2014. All entries were sown in replicated trials following the layout of an alpha design with three replications per location. Field trials were conducted at two research stations from the University of Hohenheim, Heidfeldhof (HOH) near Stuttgart-Hohenheim (latitude 48°42′38.6274″N, longitude 9°11′21.2352″E, altitude 390 m, 697 mm mean annual precipitation, 8.8 °C mean annual temperature, loess-derived stagnic Luvisol with silty loam-texture) and Oberer Lindenhof (OLI) located on the Swabian Alb (latitude 48°28′38.8524″N, longitude 9°18′52.9986″E, altitude 700 m, 952 mm mean annual precipitation, 6.6 °C mean annual temperature, Cambisol with silty loam-texture). All entries were treated according to best agronomic practices including fungicides, which however were not effective to Fusarium head blight control. Fertilizer doses were reduced to avoid lodging and possible bias on Fusarium head blight symptoms observation (Boeven et al. 2016b). To provide a uniform and high disease pressure all entries were inoculated at mid-anthesis (BBCH 65), using a small plot sprayer applying 1000 L ha−1 water with Fusarium culmorum isolate, strain Fc 46, in a concentration of 7.5 × 105 spores mL−1. To ensure similar plant development stage during inoculation, the entries were inoculated three times (Boeven et al. 2016b).
The following traits were assessed in this study: (1) Fusarium head blight symptoms were assessed on a percentage scale in several subsequent visual scorings on plot basis; (2) flowering time was determined as the number of days after January 1st to the day when 75% of the plants of a single plot were at the stage of full flowering (BBCH 65, 50% of anthers mature); (3) mean plant height in cm of each row was measured from the bottom to the erected ear at the growth stage of dough development of kernels. In particular, the first FHB scoring was assessed 20 days after inoculation, on a percentage scale. Each plot was evaluated four to five times at two-to-three day intervals. The arithmetic mean of the four or five visual scorings was used for further calculations and is referred as the mean FHB severity. Entry “2079-N” was not rated for FHB due to very late flowering.
Phenotypic data analysis
Best linear unbiased estimators (BLUEs) were adopted from a previous study conducted by Boeven et al. (2016b). A covariate adjustment for flowering time was performed by Boeven et al. (2016b) according to Emrich et al. (2008). In this manuscript, the corrected mean FHB rating for flowering time is referred to as “FHB” and the non-corrected rating as “FHBnc”. BLUEs of the 133 lines were used to calculate Pearson’s correlation (r) between traits. The significance between traits was tested with tabulated values based on the “z” transformation (Fisher 1921).
Variance components for our subset of lines were estimated in a linear mixed model framework treating genotype as random using the following model:
where E, G, R and B denote the effects of the environment, genotype, replicates and incomplete blocks, respectively. Dummy variables were used to separate the 133 genotypes from the 434 full set of genotypes and, for the sake of simplicity, were not described in the above formula (Piepho et al. 2006). Heterogeneous error variances were assumed for each location. Mixed model analysis was performed with the software package ASReml-R 3.0 (Gilmour et al. 2009)
Entry-mean heritability (h2) for the set of 133 lines was calculated using the modified formula from Hallauer et al. (2010):
where \(\hat{\sigma }_{G}^{2}\) is the estimated genotypic variance, \(\hat{\sigma }_{G \times E}^{2}\) the estimated genotype × environment interaction variance, E the number of environments, \(\hat{\sigma }_{e}^{2}\) the pooled error variance, and R the number of replicates per environment. All statistical analyses were performed within the R environment (R Core Team 2017).
Molecular data analysis
Leaf samples were collected and sent to Diversity Arrays Technology Pty. Ltd. (Canberra, Australia, www.diversityarrays.com) for genotyping. In total 105,275 markers (73,346 DArTs and 31,929 SNPs) were available. The 12% missing marker values were imputed using the software LinkImpute, based on LD-kNNi algorithm, nearest neighbor genotype imputation method with 97% imputation accuracy (Money et al. 2015). Quality control excluded markers with ≤ 5% minor allele frequency (MAF) or genotypes with more than 25% missing values. After this quality control, 66,416 markers (49,337 DArTs and 17,079 SNPs, given the letters “D” and “S” as prefix, respectively) and 123 individuals remained. Among the remaining markers, 39,913 markers had a genetic map position (Losert et al. 2017a). Both unmapped and mapped markers were used for further analysis.
Association mapping and genomic prediction
Marker-trait association analysis was computed through a GWA using the R package GenABEL (Aulchenko et al. 2007). Principal component analysis (PCA) did not detect any noticeable population structure (Suppl. Figure 1), and thus a kinship matrix (K) was included in the mixed model to account for possible population stratification in this data set (Lander and Schork 1994; Yu et al. 2006; Würschum 2012). Association mapping was performed with both mapped and unmapped imputed markers, and the phenotypic trait data. The most likely chromosomal position of the significant unmapped markers was obtained from their linkage disequilibrium with the mapped markers. Fixed marker effects were tested for significance by obtaining the P value from the marker-trait association within the function “mmscore” in GenABEL (Aulchenko et al. 2007). To control for multiple testing, the threshold for significant marker-trait associations was set to P < 0.0001. After correction for collinearity, the total proportion of explained genotypic variance (pG) of the significant marker-trait associations was estimated by the ratio \({p_{G}}= \frac{{R_{adj}^{2} }}{{h^{2} }}\) (Utz et al. 2000). The adjusted \(R^{2} = (R_{adj}^{2} )\) was computed by fitting all significant QTL simultaneously in a linear model, in decreasing order of marker-trait association strength. The explained genotypic variance for each QTL was derived from the sum of squares of that QTL in the linear model (Würschum et al. 2015). The additive effect (α-effect) of each significant marker was estimated with a linear model fitting only one significant QTL at a time.
Genomic prediction was carried out by ridge-regression BLUP (RR-BLUP; Whittaker et al. 2000) with the R package “rrBLUP” (Endelman 2011; Endelman and Jannink 2012) using mapped and unmapped imputed markers. We also performed a weighted ridge-regression BLUP (wRR-BLUP) where significant markers with more than 5% explained genotypic variance detected in the GWA were used as fixed effect in the genomic prediction model (Zhao et al. 2014; Spindel et al. 2016). The prediction accuracy was defined as the Pearson’s correlation coefficient between observed and predicted trait values divided by the square root of the trait heritability.
Results
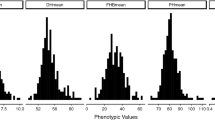
The genotypic variance \(\left( {\hat{\sigma }_{G}^{2} } \right)\) as well as the genotype-by-environment interaction variance \(\left( {\hat{\sigma }_{G \times E}^{2} } \right)\) were significantly different from zero (P < 0.001) for all evaluated traits. Adjusted entry means across four environments revealed 14.76 to 70.70% FHB damage in our set of 133 triticale lines (Table 1). The distribution of these lines had a slight shift towards resistant entries (Fig. 1). The lines “Cultivo” and “0242-N” were the most and least resistant to FHB, respectively. Plant height also showed a significant large variation ranging from 81.20 to 137.37 cm, with “1692-N” from Switzerland and “Balu PZO” from Germany being the shortest and tallest genotypes, respectively (Suppl. Table 1). All traits showed high entry-mean heritability estimates (> 0.8), underscoring the high quality of the phenotypic data.

Frequency distribution, phenotypic correlation coefficients and scatter plots for and between the evaluated traits. FHBnc, visual scoring of Fusarium head blight not corrected for flowering time; FHB, visual scoring of Fusarium head blight corrected for flowering time; FT, flowering time; PH plant height. *, *** Significantly different from zero at 0.05, and 0.001 level of probability, respectively
A moderate negative correlation between non-corrected FHB ratings (FHBnc) and flowering time of r = − 0.39 (P < 0.001) was found. After correction for flowering time, this correlation was no longer significant (r = − 0.11, P = 0.22). The following analyses were therefore carried out with the corrected FHB values. The correlation between plant height and FHB was significant, but rather low (r = 0.18, P = 0.048).
Genome-wide association mapping identified six QTL for FHB, three for flowering time, and three for plant height (Table 2). For FHB, one QTL was identified on chromosome 2A, two on chromosomes 2B and 5B, and one on chromosome 3R (Fig. 2). The proportion of genotypic variance explained by these QTL ranged from 0.28 to 30.23%. A major QTL was identified on chromosome 5B, explaining 30.23% of the genotypic variance and another on chromosome 3R, explaining 15.38% of the genotypic variance. For flowering time, the genome-wide scan revealed QTL on chromosomes 3A, 6B and 6R, individually explaining 4.58 to 15.64% of the genotypic variance. Plant height QTL were identified on chromosomes 2A, 4B and 4R, explaining 1.26 to 16.25% of the genotypic variance. The total explained variance was 56.64, 27.70 and 19.87% for FHB, flowering time and plant height, respectively.

Manhattan plot of the genome-wide association study of Fusarium head blight (FHB) severity. The significance threshold (P < 0.0001) is indicated by the dashed horizontal line
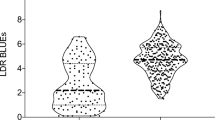
The allelic classes at the FHB QTL with ≥ 5% explained genotypic variance showed considerable differences in FHB severity. Interestingly, the majority of the triticale lines already carried the resistance-conferring alleles (Fig. 3). Consistently, the most resistant triticale lines combined the resistance alleles of the four most important QTL (Suppl. Table 1).

Box plots for FHB severity. Notched boxplots of QTL with ≥ 5% explained genotypic variance for Fusarium head blight (FHB) severity. If notches do not overlap, this indicates that medians are different according to McGill et al. (1978) at P < 0.05; horizontal line within boxes = median, and white circle = outliers; the numbers (n) below the boxplots indicate the number of triticale lines bearing the respective allele
In addition, we assessed the accuracy of genome-wide prediction approaches, RR-BLUP and wRR-BLUP (Fig. 4). The prediction accuracy achieved with RR-BLUP ranged from 0.37 to 0.57. Interestingly, wRR-BLUP enabled a higher prediction accuracy than RR-BLUP for all three traits, varying from 0.52 for plant height, 0.71 for flowering time to 0.78 for FHB resistance (Fig. 4).

Prediction accuracy for Fusarium head blight (FHB) severity, flowering time (FT) and plant height (PH) based on genome-wide prediction after cross validation with ridge-regression BLUP (RR-BLUP), and weighted ridge-regression BLUP (wRR- BLUP) including the QTL explaining more than 5% of the genotypic variance for each trait as fixed effects in the model
Discussion
Improving complex traits by phenotypic selection is often laborious and time-consuming, and for FHB the resistance test has to be performed during flowering and ripening thus requiring an entire crop season. The aim of this study was to evaluate the potential of genomic tools to assist FHB resistance breeding.
Variation for FHB resistance can be exploited in triticale breeding
The 133 winter triticale lines included in this study showed a large variation for FHB resistance, illustrating that improvement through selection is feasible. The genotype “Lasko”, for example, previously known as the most resistant cultivar to FHB (Oettler and Wahle 2001), again showed an above-average FHB resistance. However, this rather old genotype, released in 1986, is of little interest for current breeding programs, as it now has a comparably low agronomic performance. More interesting are recent, resistant cultivars like “Adverdo” and “Grenado” that were widely grown in Germany in 2013 and 2014 (Bundessortenamt 2014) and that combine both resistance against FHB and good agronomic performance (Suppl. Table 1).
We observed a negative correlation between not-corrected FHB and flowering time as already reported in many other studies of small-grain cereals (Mesterházy 1995; Miedaner 1997; Buerstmayr et al. 2000; Emrich et al. 2008; Schmolke et al. 2005; Kalih et al. 2014; Miedaner et al. 2017). To provide a more helpful ranking of genotypes to the breeder and to avoid a co-selection for late maturity, a covariate adjustment for flowering time appears appropriate and can be recommended also in triticale (Emrich et al. 2008).
Besides flowering time, plant height can also have an influence on FHB resistance (Mesterházy 1995; Miedaner and Voss 2008; Becher et al. 2013; Kalih et al. 2014; Miedaner et al. 2017). Especially when reduced height genes are present, the FHB susceptibility tends to increase in wheat (Miedaner and Voss 2008; Becher et al. 2013) as well as in triticale (Kalih et al. 2014, 2015). Although a positive correlation between grain yield and plant height was reported by Losert et al. (2017b), a limitation of plant height is important for breeding as it reduces the risk of lodging (Liu et al. 2015). In our study, no significant correlation between plant height and FHB was observed. This might be due to the composition of the triticale panel comprising elite lines that have already been selected for agronomic traits and indicates that lines with reduced height could be selected from this panel without increasing FHB susceptibility. Accordingly, the cultivar “Cultivo” showed the highest FHB resistance and a plant height below average (Suppl. Table 1). However, “Cultivo” is a rather late variety and it is possible that our disease assessment system was favoring late genotypes even after correction for flowering time (Boeven et al. 2016b).
Inheritance of plant height and flowering time
We identified three QTL for plant height, but no QTL was detected on chromosome 5R, that has previously been shown to harbor the major height-reducing locus Ddw1 (Kalih et al. 2014). While cultivars known to carry Ddw1 were included in this study, this might be due to a low frequency of this dwarfing locus in our panel, as the power to detect rare alleles in GWA is low (van Inghelandt et al. 2011; Würschum 2012).
Flowering time is an important adaptive trait for small-grain cereals (Passioura 1996; Slafer and Whitechurch 2001; Hackauf et al. 2017). Our study identified three QTL for flowering time that were not overlapping with FHB QTL. This confirms the successful correction for the confounding effects of flowering time on FHB resistance and indicates that FHB QTL can be identified with the corrected trait data.
Genetic architecture of FHB resistance in triticale
For FHB resistance, we detected six QTL that individually explained a proportion of genotypic variance between 0.28 and 30.23%. While two QTL explained more than 10% of the genotypic variance, this nevertheless corroborates previous findings, illustrating that, as in other small-grain cereals, FHB resistance in triticale is a complex trait. This conclusion was supported by the genome-wide prediction approach, which suggested additional small-effect QTL to be involved in the genetic control of FHB resistance. Kalih et al. (2014) also identified several minor and a few major QTL conferring resistance to FHB in triticale. However, their higher explained genotypic variance of 58.60% was mainly caused by the rye dwarfing gene Ddw1 on chromosome 5R, where lines with the dwarfing allele were significantly more susceptible. In a different study, Kalih et al. (2015) also identified some major QTL in each of four triticale families. Dhariwal et al. (2018) reported 17 QTL for percentage of ears infected (DI), percentage of spikelets infected (DS), VRI (DI × DS/100) and DON content in one double-haploid population between the spring triticale line “TMP16315” and a Canadian triticale cultivar “AC Ultima”. In this population, individual QTL varying from 2.9 to 34.1% explained phenotypic variance were detected, that were also not co-located with dwarfing genes.
In our study, we identified a major QTL on chromosome 5B explaining 30.23% of the genotypic variance. Kalih et al. (2015) also identified a QTL on the same chromosome in one of the four evaluated populations, but the different marker systems used in the two studies prevent a direct comparison. Furthermore, we also identified a major QTL for FHB resistance on chromosome 3R that explained 15.38% of the genotypic variance and has not been reported before. Given a positive validation of this resistance QTL in different genetic backgrounds, it can be exploited as a new source of FHB resistance in triticale. Interestingly, the 94 lines already combined the resistance alleles of four of the QTL with explained genetic variance above 5% (Suppl. table 1). Nevertheless, due to the complexity of FHB genetic architecture we encourage further studies with larger population sizes in order to increase the QTL detection power and therefore, increase the likelihood of identifying additional QTL with low explained variance. For the identified QTL not the resistance-conferring alleles appear to be rare, but the susceptible alleles, which might explain the overall higher Fusarium resistance of triticale compared to bread wheat.
Prospects of genomics-assisted breeding
In our study, we observed a high heritability of 0.81 for FHB severity, which is usually not achievable in commercial breeding programs where FHB severity is tested among thousands of entries in unreplicated trials. However, our results and the high genotype-by-environment interaction suggest that also for FHB selection in breeding, multi-environmental field trials are recommendable. Moreover, recent stripe rust epidemics can lead to a confounding effect when this disease is also present on triticale ears in FHB trials (Miedaner et al. 2004; Losert et al. 2017a). These challenges might be tackled with the aid of genomic tools.
Our results clearly illustrate the genetic complexity of FHB resistance in triticale. Marker-assisted selection (MAS) is a genomic tool that can be utilized if a few medium- to large-effect QTL are responsible for a trait. Thus, only few of the identified FHB QTL may hold potential for MAS. Importantly, their effects need to be validated in different breeding material before they can be applied more widely. Experience for MAS of FHB QTL in wheat has shown, that these QTL are often background dependent and consequently, the success of MAS to improve FHB resistance in triticale appears limited (Jiang et al. 2017).
Genomic selection is a promising alternative to assist breeding for improved FHB resistance. The potential of GS in triticale has already been demonstrated for several agronomic traits (Würschum et al. 2017). In our study we obtained mean cross-validated prediction accuracies of 0.55, 0.57, and 0.37 for FHB resistance, flowering time, and plant height, respectively. Interestingly, weighted RR-BLUP substantially increased the prediction accuracy from 0.55 up to 0.78 for FHB resistance, which is in line with previous studies in wheat and triticale (Zhao et al. 2014; Boeven et al. 2016a; Losert et al. 2017a).
Taken together, the few identified medium-effect QTL might be employed in marker-assisted selection, provided their effects can be confirmed in other breeding material. The accuracy of genomic prediction for FHB in triticale was promising, illustrating the potential of this approach to increase the selection gain for Fusarium resistance in triticale breeding programs.
Implementation of GS in triticale breeding programs
In the following, we discuss a possible implementation of GS for FHB resistance in triticale breeding programs. After crossing, early generation lines are grown as a single row. Inoculation of these plots is not feasible because FHB heavily impairs seed germination and quality. Thus, GS displays an attractive option to select for FHB resistance at this early stage where seed availability is a bottleneck. The implementation of GS is possible with historical data when a training population with genotypic and good phenotypic data is available. The relatedness between training and the actual breeding population is, however, an important parameter for a successful GS (Gowda et al. 2014). However, the composition of the training population and how to implement GS in breeding programs is still a field of intensive research. The training population is then used to predict the genomic estimated breeding values (GEBVs) of the breeding population (Bassi et al. 2016), based on which a negative selection of the least performant candidates can already take place. This liberal FHB selection allows enough remaining variation for other traits of interest such as grain yield, other agronomic or other disease resistance traits. When the breeding program has a high priority for FHB resistance, a higher selection intensity can also be implemented requiring, however, a larger population size. The candidates that make it to the first yield trial are phenotyped for FHB resistance to confirm their resistance level and simultaneously update the training population for the next breeding cycle. The indirect selection with genomic data already in the early stages of a breeding program can be expected to lead to a shift of the population mean towards higher Fusarium resistance.
In general, the decision of whether to employ phenotypic or genomic selection for FHB depends on the prediction accuracy and the availability of genotypic data. If marker data are available anyhow, they can also be used for improving FHB resistance and other disease resistances. If for economic reasons GS is only employed at later stages, e.g. in parallel to the first yield trials, it can still be valuable to complement the phenotypic data and thus improve the FHB rating.
Conclusions
Taken together, our analysis revealed a substantial genetic variation for FHB resistance in triticale that can be exploited for a further improvement of the resistance level in breeding germplasm. The first genome-wide association study for FHB resistance in triticale identified two major and several minor QTL, thus confirming the complex genetic architecture of this resistance. After validation of the major QTL, they could be utilized in triticale breeding through marker-assisted selection. In addition, a promising prediction accuracy was obtained for genomic selection, illustrating that it can be a valuable tool for triticale disease resistance breeding. Nevertheless, our results also underscore the value of field phenotyping to obtain robust and reliable phenotypic data. Thus, genomic approaches have the potential to assist breeding of high yielding material with high-level disease resistance, an important criterion to maintain the importance of triticale in the seed market.
Abbreviations
- BLUEs:
-
Best linear unbiased estimators
- DON:
-
Deoxynivalenol
- FHB:
-
Fusarium head blight
- FT:
-
Flowering time
- GEBV:
-
Genomic estimated breeding value
- GS:
-
Genomic selection
- GWA:
-
Genome-wide association
- K:
-
Kinship
- MAF:
-
Minor allele frequency
- MAS:
-
Marker-assisted selection
- NIV:
-
Nivalenol
- PCA:
-
Principal component analysis
- PH:
-
Plant height
- QTL:
-
Quantitative trait loci
- RR-BLUP:
-
Ridge-regression BLUP
- wRR-BLUP:
-
Weighted ridge-regression BLUP
- ZON:
-
Zearalenone
References
Audenaert K, Vanheule A, Höfte M, Haesaert G (2013) Deoxynivalenol: a major player in the multifaceted response of Fusarium to its environment. Toxins 6:1–19. https://doi.org/10.3390/toxins6010001
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: an R library for genome-wide association analysis. Bioinformatics 23:1294–1296. https://doi.org/10.1093/bioinformatics/btm108
Bassi FM, Bentley AR, Charmet G et al (2016) Breeding schemes for the implementation of genomic selection in wheat (Triticum spp.). Plant Sci 242:23–36. https://doi.org/10.1016/j.plantsci.2015.08.021
Becher R, Miedaner T, Wirsel SGR (2013) Biology, diversity, and management of FHB-causing Fusarium species in small-grain cereals. In: Kempken F (ed) Agricultural applications. 2nd Edition. The Mycota XI. Springer, Berlin, pp 199–241
Bennett JW, Klich M (2003) Mycotoxins. Clin Microbiol Rev 16:497–516. https://doi.org/10.1128/CMR.16.3.497
Bird SH, Rowe JB, Choct M et al (1999) In vitro fermentation of grain and enzymatic digestion of cereal starch. Recent Adv Anim Nutr Aust 12:53–62
Boeven PHG, Longin CFH, Leiser WL et al (2016a) Genetic architecture of male floral traits required for hybrid wheat breeding. Theor Appl Genet 129:2343–2357. https://doi.org/10.1007/s00122-016-2771-6
Boeven PHG, Würschum T, Weissmann S et al (2016b) Prediction of hybrid performance for Fusarium head blight resistance in triticale (× Triticosecale Wittmack). Euphytica 207:475–490. https://doi.org/10.1007/s10681-015-1498-9
Bolduan C, Miedaner T, Schipprack W et al (2009) Genetic variation for resistance to ear rots and mycotoxins contamination in early European maize inbred lines. Crop Sci 49:2019–2028. https://doi.org/10.2135/cropsci2008.12.0701
Buerstmayr H, Steiner B, Lemmens M, Ruckenbauer P (2000) Resistance to Fusarium head blight in winter wheat: heritability and trait associations. Crop Sci 40:1012–1018. https://doi.org/10.2135/cropsci2000.4041012x
Bundessortenamt (2014) Beschreibende Sortenliste: Getreide, Mais, Öl- und Faserpflanzen, Leguminosen, Rüben, Zwischenfrüchte. http://www.bundessortenamt.de/internet30/file%0Aadmin/Files/PDF/bsl_getreide_2014.pdf%0A. Accessed 9th Aug 2018
Čonková E, Laciaková A, Kováč G, Seidel H (2003) Fusarial toxins and their role in animal diseases. Vet J 165:214–220. https://doi.org/10.1016/S1090-0233(02)00127-2
Dhariwal R, Fedak G, Dion Y et al (2018) High density single nucleotide polymorphism (SNP) mapping and quantitative trait loci (QTL) analysis in a biparental spring triticale population localized major and minor effect Fusarium head blight resistance and associated traits QTL. Genes 9:1–26. https://doi.org/10.3390/genes9010019
EFSA (2004) Opinion of the scientific panel on contaminants in the food chain on a request from the commission related to deoxynivalenol (DON) as undesirable substance in animal feed. EFSA J 73:1–41
Emrich K, Wilde F, Miedaner T, Piepho HP (2008) REML approach for adjusting the Fusarium head blight rating to a phenological date in inoculated selection experiments of wheat. Theor Appl Genet 117:65–73. https://doi.org/10.1007/s00122-008-0753-z
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome J 4:250–255. https://doi.org/10.3835/plantgenome2011.08.0024
Endelman JB, Jannink J (2012) Shrinkage estimation of the realized relationship matrix. G3 Genes Genomes Genet 2:1405–1413. https://doi.org/10.1534/g3.112.004259
European Commission (2006) Commission Recommendation (EC) No 576/2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 and HT-2 and fumonisins in products intended for animal feeding. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri = OJ:L:2006:229:0007:0009:EN:PDF. Accessed on 23rd March 2015
FAOSTAT (2018) Statistical databases and datasets of the food and agriculture organization of the United Nations. http://www.fao.org/faostat/en/#data/QC. Accessed 5th Feb 2018
Fisher RA (1921) On the “probable error” of a coefficient of correlation deduced from a small sample. Metron 1:3–32
Gilmour AR, Gogel BJ, Cullis BR, Thompson R (2009) ASReml user guide release 3.0. VSN International Ltd, Hemel Hempstead, p 275
Gowda M, Hahn V, Reif JC et al (2011) Potential for simultaneous improvement of grain and biomass yield in Central European winter triticale germplasm. Field Crops Res 121:153–157. https://doi.org/10.1016/j.fcr.2010.12.003
Gowda M, Zhao Y, Würschum T, Longin CF et al (2014) Relatedness severely impacts accuracy of marker-assisted selection for disease resistance in hybrid wheat. Heredity 112:552–561. https://doi.org/10.1038/hdy.2013.139
Hackauf B, Haffke S, Fromme FJ et al (2017) QTL mapping and comparative genome analysis of agronomic traits including grain yield in winter rye. Theor Appl Genet 130:1801–1817. https://doi.org/10.1007/s00122-017-2926-0
Hallauer AR, Carena MJ, Filho J (2010) Quantitative genetics in maize breeding, 3rd edn. Handbook of plant breeding. Springer, New York
Heffner EL, Lorenz AJ, Jannink JL, Sorrells ME (2010) Plant breeding with Genomic selection: gain per unit time and cost. Crop Sci 50:1681–1690. https://doi.org/10.2135/cropsci2009.11.0662
Jiang Y, Schulthess AW, Rodemann B et al (2017) Validating the prediction accuracies of marker-assisted and genomic selection of Fusarium head blight resistance in wheat using an independent sample. Theor Appl Genet 130:471–482. https://doi.org/10.1007/s00122-016-2827-7
Kalih R, Maurer HP, Hackauf B, Miedaner T (2014) Effect of a rye dwarfing gene on plant height, heading stage, and Fusarium head blight in triticale (× Triticosecale Wittmack). Theor Appl Genet 127:1527–1536. https://doi.org/10.1007/s00122-014-2316-9
Kalih R, Maurer HP, Miedaner T (2015) Genetic architecture of fusarium head blight resistance in four winter triticale populations. Phytopathology 105:334–341. https://doi.org/10.1094/PHYTO-04-14-0124-R
Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048
Liu W, Leiser WL, Maurer HP et al (2015) Evaluation of genomic approaches for marker-based improvement of lodging tolerance in triticale. Plant Breed 134:416–422
Löffler M, Schön CC, Miedaner T (2009) Revealing the genetic architecture of FHB resistance in hexaploid wheat (Triticum aestivum L.) by QTL meta-analysis. Mol Breed 23:473–488. https://doi.org/10.1007/s11032-008-9250-y
Losert D, Maurer HP, Leiser WL, Würschum T (2017a) Defeating the Warrior: genetic architecture of triticale resistance against a novel aggressive yellow rust race. Theor Appl Genet 130:685–696. https://doi.org/10.1007/s00122-016-2843-7
Losert D, Maurer HP, Marulanda JJ, Würschum T (2017b) Phenotypic and genotypic analyses of diversity and breeding progress in European triticale (× Triticosecale Wittmack). Plant Breed 136:18–27. https://doi.org/10.1111/pbr.12433
Martin M, Dhillon BS, Miedaner T, Melchinger AE (2012) Inheritance of resistance to Gibberella ear rot and deoxynivalenol contamination in five flint maize crosses. Plant Breed 131:28–32. https://doi.org/10.1111/j.1439-0523.2011.01908.x
McGill R, Tukey JW, Larsen WA (1978) Variations of box plots. Am Stat 32(1):12–16
Mergoum M, Singh PK, Peña RJ et al (2009) Triticale: a ‘new’ crop with old challenges. In: Carena MJ (ed) Cereals. Handbook of plant breeding, vol 3. Springer, New York, NY, pp 267–287. https://doi.org/10.1007/978-0-387-72297-9_9
Mesterházy A (1995) Types and components of resistance to Fusarium head blight of wheat. Plant Breed 114:377–386. https://doi.org/10.1111/j.1439-0523.1995.tb00816.x
Miedaner T (1997) Breeding wheat and rye for resistance to Fusarium diseases. Plant Breed 116:201–220. https://doi.org/10.1111/j.1439-0523.1997.tb00985.x
Miedaner T, Voss HH (2008) Effect of dwarfing Rht genes on fusarium head blight resistance in two sets of near-isogenic lines of wheat and check cultivars. Crop Sci 48:2115–2122. https://doi.org/10.2135/cropsci2008.02.0107
Miedaner T, Heinrich N, Schneider B et al (2004) Estimation of deoxynivalenol (DON) content by symptom rating and exoantigen content for resistance selection in wheat and triticale. Euphytica 139:123–132. https://doi.org/10.1007/s10681-004-2489-4
Miedaner T, Caixeta F, Talas F (2013) Head-blighting populations of Fusarium culmorum from Germany, Russia, and Syria analyzed by microsatellite markers show a recombining structure. Eur J Plant Pathol 137:743–752. https://doi.org/10.1007/s10658-013-0284-6
Miedaner T, Kalih R, Großmann MS, Maurer HP (2016) Correlation between Fusarium head blight severity and DON content in triticale as revealed by phenotypic and molecular data. Plant Breed 135:31–37. https://doi.org/10.1111/pbr.12327
Miedaner T, Sieber AN, Desaint H et al (2017) The potential of genomic-assisted breeding to improve Fusarium head blight resistance in winter durum wheat. Plant Breed 136:610–619. https://doi.org/10.1111/pbr.12515
Mirdita V, He S, Zhao Y, Korzun V et al (2015) Potential and limits of whole genome prediction of resistance to Fusarium head blight and Septoria tritici blotch in a vast Central European elite winter wheat population. Theor Appl Genet 128:2471–2481. https://doi.org/10.1007/s00122-015-2602-1
Money D, Gardner K, Migicovsky Z et al (2015) LinkImpute: fast and accurate genotype imputation for nonmodel organisms. Genes Genomes Genet 5:2383–2390. https://doi.org/10.1534/g3.115.021667
Oettler G (2005) The fortune of a botanical curiosity—triticale: past, present and future. J Agric Sci 143:329–346. https://doi.org/10.1017/S0021859605005290
Oettler G, Wahle G (2001) Genotypic and environmental variation of resistance to head blight in triticale inoculated with Fusarium culmorum. Plant Breed 120:297–300. https://doi.org/10.1046/j.1439-0523.2001.00611.x
Oettler G, Heinrich N, Miedaner T (2004) Estimates of additive and dominance effects for Fusarium head blight resistance of winter triticale. Plant Breed 123:525–530
Passioura JB (1996) Drought and drought tolerance. Plant Growth Regul 20:79–83. https://doi.org/10.1007/978-94-017-1299-6_1
Piepho HP, Williams ER, Fleck M (2006) A note on the analysis of designed experiments with complex treatment structure. HortScience 41:446–452
Pronyk C, Mazza G (2011) Optimization of processing conditions for the fractionation of triticale straw using pressurized low polarity water. Biores Technol 102:2016–2025. https://doi.org/10.1016/j.biortech.2010.09.064
Scherm B, Balmas V, Spanu F et al (2013) Fusarium culmorum: causal agent of foot and root rot and head blight on wheat. Mol Plant Pathol 14:323–341. https://doi.org/10.1111/mpp.12011
Schmolke M, Zimmermann G, Buerstmayr H et al (2005) Molecular mapping of Fusarium head blight resistance in the winter wheat population Dream/Lynx. Theor Appl Genet 111:747–756. https://doi.org/10.1007/s00122-005-2060-2
Slafer GA, Whitechurch EM (2001) Manipulating wheat development to improve adaptation and to search for alternative opportunities to increase yield potential. In: Reynolds MP, Ortiz-Monasterio JI, McNab A (eds) Application of physiology in wheat breeding. CIMMYT, Mexico, pp 160–170
Spindel JE, Begum H, Akdemir D et al (2016) Genome-wide prediction models that incorporate de novo GWA are a powerful new tool for tropical rice improvement. Heredity 116:395–408. https://doi.org/10.1038/hdy.2015.113
Sugita-Konishi Y, Kubosaki A, Takahashi M et al (2008) Nivalenol and the targeting of the female reproductive system as well as haematopoietic and immune systems in rats after 90-day exposure through the diet. Food Addit Contam Part A Chem Anal Control Exposure Risk Assess 25:1118–1127. https://doi.org/10.1080/02652030802093892
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing
Utz HF, Melchinger AE, Schön CC (2000) Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154:1839–1849. https://doi.org/10.2307/1403680
van Inghelandt D, Reif JC, Dhillon BS et al (2011) Extent and genome-wide distribution of linkage disequilibrium in commercial maize germplasm. Theor Appl Genet 123:11–20. https://doi.org/10.1007/s00122-011-1562-3
Whittaker JC, Thompson R, Denham MC (2000) Marker-assisted selection using ridge regression. Genet Res 75:249–252
Würschum T (2012) Mapping QTL for agronomic traits in breeding populations. Theor Appl Genet 125:201–210. https://doi.org/10.1007/s00122-012-1887-6
Würschum T, Langer SM, Longin CFH (2015) Genetic control of plant height in European winter wheat cultivars. Theor Appl Genet 128:865–874. https://doi.org/10.1007/s00122-015-2476-2
Würschum T, Maurer HP, Weissmann S et al (2017) Accuracy of within- and among-family genomic prediction in triticale. Plant Breed 136:230–236. https://doi.org/10.1111/pbr.12465
Yu J, Pressoir G, Briggs WH et al (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208. https://doi.org/10.1038/ng1702
Zhao Y, Mette MF, Gowda M et al (2014) Bridging the gap between marker-assisted and genomic selection of heading time and plant height in hybrid wheat. Heredity 112:638–645. https://doi.org/10.1038/hdy.2014.1
Acknowledgements
The molecular marker data was funded by the Federal Ministry of Food and Agriculture (BMEL) through its project management body Fachagentur für Nachwachsende Rohstoffe e.V. (FNR) (Grants: 22406112, 22406212, 22406312, and 22406412). We thank the Federal Ministry of Food and Agriculture (BMEL) based on a decision of the Parliament of the Federal Republic of Germany via the Federal Office for Agriculture and Food (BLE) under the innovation support program within the PRIMA cooperative project (Grant No. 2818202815) for financially supporting the first author of this project. The authors also would like to thank Tizian Zollinger for the collection of phenotypic data within his master thesis and Dr. S. Weissmann, HegeSaat GmbH & Co. KG, Singen, for providing genotypes. We highly appreciate the excellent technical support of the teams at Hohenheim and Oberer Lindenhof.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
The authors declare that the experiments comply with the current laws of Germany.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Galiano-Carneiro, A.L., Boeven, P.H.G., Maurer, H.P. et al. Genome-wide association study for an efficient selection of Fusarium head blight resistance in winter triticale. Euphytica 215, 4 (2019). https://doi.org/10.1007/s10681-018-2327-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-018-2327-8