
Abstract
Key message
Compared with independent validation, cross-validation simultaneously sampling genotypes and environments provided similar estimates of accuracy for genomic selection, but inflated estimates for marker-assisted selection.
Abstract
Estimates of prediction accuracy of marker-assisted (MAS) and genomic selection (GS) require validations. The main goal of our study was to compare the prediction accuracies of MAS and GS validated in an independent sample with results obtained from fivefold cross-validation using genomic and phenotypic data for Fusarium head blight resistance in wheat. In addition, the applicability of the reliability criterion, a concept originally developed in the context of classic animal breeding and GS, was explored for MAS. We observed that prediction accuracies of MAS were overestimated by 127% using cross-validation sampling genotype and environments in contrast to independent validation. In contrast, prediction accuracies of GS determined in independent samples are similar to those estimated with cross-validation sampling genotype and environments. This can be explained by small population differentiation between the training and validation sets in our study. For European wheat breeding, which is so far characterized by a slow temporal dynamic in allele frequencies, this assumption seems to be realistic. Thus, GS models used to improve European wheat populations are expected to possess a long-lasting validity. Since quantitative trait loci information can be exploited more precisely if the predicted genotype is more related to the training population, the reliability criterion is also a valuable tool to judge the level of prediction accuracy of individual genotypes in MAS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wheat is an important staple crop providing one-fifth of the total calories of the world’s population (Reynolds et al. 2011). Wheat grain yield and quality is severely impacted by Fusarium head blight (FHB) (Buerstmayr et al. 2009). FHB infection in wheat production cannot be completely controlled by fungicide application, crop rotation, and soil tillage alone (Paul et al. 2010) entailing the need to breed and grow wheat varieties which are resistant against FHB (Miedaner et al. 2011).
Genomic-assisted breeding can potentially accelerate selection gain for FHB resistance (Miedaner et al. 2009, 2011; Mirdita et al. 2015a, b). Two powerful genomic-assisted breeding tools are marker-assisted (Lande and Thompson 1990) and genomic selection (Meuwissen et al. 2001). In marker-assisted selection (MAS), the performance of individuals is estimated using a few functional markers. In contrast, in genome-wide selection (GS) the trait performance is predicted using many markers without performing marker-specific significance tests (Zhao et al. 2015a).
The efficiency of MAS and GS depend on many factors such as the genetic architecture of the target trait as well as the genetic composition of the population used to estimate the marker effects (Heffner et al. 2009; Zhao et al. 2015b). Therefore, prediction accuracies of MAS and GS have to be estimated in order to optimally use genomics in breeding programs. The estimates of prediction accuracies should rely on relevant germplasm and have to be validated either via cross-validation (Hjorth 1994) or by validation with independent samples (e.g., Melchinger et al. 1998). Here “samples” means both genotypes and environments where the genotypes are tested, while “independent” means that samples are independently sampled from a certain population. This is relevant for breeding as the lines entering a breeding program are typically derived from a pool of elite parental genotypes, and the target of breeding is to select genotypes that performed well in a target population of environments (TPE, Atlin et al. 2000).
The main difference between cross-validation and validation with independent samples is that the latter involves prediction for untested genotypes in untested environments. Recently, this problem has been studied using cross-validation sampling genotypes and environments (Jarquin et al. 2014; Malosetti et al. 2016; Saint Pierre et al. 2016). However, these studies focused on predicting the performances of genotypes within environments. This is relevant when the genotypes strongly interact with environments, in which case it is unrealistic to set the target of breeding across all environments. In contrast, if the genotype-by-environment interaction is moderate, it makes more sense to treat the environments as a sample from a TPE and predict the genotypes across environments. Our study focused on the latter case.
Most previous studies on the potential and limits of MAS and GS of FHB resistance in wheat rely on cross-validation (Arruda et al. 2015; Jiang et al. 2015; Mirdita et al. 2015a, b; Rutkoski et al. 2012). Validation using independent samples is to our knowledge lacking. This is not critical for biparental populations, because prediction accuracies are comparable for cross-validation and validation with independent samples (Utz et al. 2000). Nevertheless, prediction accuracies for cross-validation and validation with independent samples may differ when considering panels of genetically diverse lines, like for example in association mapping. For diversity panels, relatedness between training and validation population impacts the prediction accuracy of both, MAS and GS (Gowda et al. 2014; Habier et al. 2007). Therefore, applying fivefold cross-validation is not necessarily reflecting the validation scenario relevant for applied plant breeding and stratified sampling may be required.
Alternatively, prediction accuracies can be estimated at the level of single genotypes rather than for entire populations. The prediction accuracy for individual genotypes is denoted as reliability and has been proposed and applied for genomic selection in the context of animal breeding (Hayes et al. 2009; Henderson 1973; VanRaden et al. 2009). The reliability criterion is not routinely applied for GS in plant breeding, despite its huge potential as recently highlighted by a pioneering study using a large wheat population (He et al. 2016). Moreover, the potential of using the reliability criterion for assessing the prediction accuracies of individual genotypes in MAS has not yet been investigated.
Our study is based on genomic and phenotypic data for FHB resistance of two independent samples of European wheat varieties released at distinct time periods. The objectives of our study were to (1) evaluate the potential and limits of MAS and GS for FHB resistance using an independent validation sample; (2) contrast the prediction accuracies of validation in an independent sample with results obtained from fivefold cross-validation, and (3) assess the potential of the reliability criterion to estimate the prediction accuracies of MAS and GS at an individual level.
Materials and methods
Plant materials
In this study we considered data sets from two experiments. The first data set (experiment I) consisted of 372 European wheat varieties which were released in the time period ranging from 1975 to 2009 (Supplementary Table S1). The varieties were evaluated for FHB resistance using multi-location field trials in Germany in 2009 and 2010 (Jiang et al. 2015; Kollers et al. 2013). In total there were four environments (location–year combinations). The second data set (experiment II) comprised 151 European wheat varieties (Supplementary Table S1) tested in three different environments in Germany in 2013 and 2014. Excluding the 18 common genotypes with experiment I, all varieties of experiment II were released between 2007 and 2010. In this study, we treated the genotypes and environments in experiments I and II as independent samples from the population of European elite wheat varieties and the TPE of Central Europe.
In both experiments, varieties were evaluated in field trials with two or three replications in a randomized complete block design. In each environment, lines were artificially spray-inoculated as described in detail by Kollers et al. (2013). FHB resistance was expressed as FHB score = FHB incidence × FHB severity/100%, where FHB incidence represents the percentage of infected spikes in a test plot and FHB severity refers to the mean percentage of infected area on infected spikes. Thus FHB score has a possible range from 0% (most resistant) to 100% (most susceptible).
Phenotypic data analyses
For both data sets, we performed one-step phenotypic analyses using linear mixed models (Smith et al. 2005). The following model was used for experiment I:
where y ijk denotes the phenotypic record of the ith genotype in the kth replication of the jth environment, μ is the common intercept term, G i is the effect of the ith genotype, E j is the effect of the jth environment, (G × E) ij is the interaction effect between the ith genotype and the jth environment, r (j)k is the effect of the kth replication in the jth environment and e ijk is the residual term which is independent and identically distributed (i.i.d) and normally distributed.
For experiment II we used a slightly different model in order to separate the 133 genotypes only in experiment II and the 18 genotypes appearing in both experiments:
where \(y_{ijk} ,\mu ,E_{j} ,r_{\left( j \right)k}\) and e are the same as before, \(\chi_{i}\) takes the value 1 if the ith genotype is only in experiment II and 0 if it is also in experiment I. The effect of the ith genotype is denoted by \(V_{i}\) (or \(T_{i}\)) if the genotype is in experiment II (or experiment I). The corresponding interaction effect with the jth environment is denoted by \(( {V \times E} )_{ij} \left( {{\text{or }} ( {T \times E} )_{ij} } \right)\).
In both models, we first assumed all effects except the intercept as random to estimate the variance components. The broad-sense heritability on an entry-mean basis was calculated as \(h^{2} = \sigma_{G}^{2} /(\sigma_{G}^{2} + \frac{{\sigma_{G \times E}^{2} }}{t} + \frac{{\sigma_{e}^{2} }}{tr})\), where \(\sigma_{G}^{2} ,\sigma_{G \times E}^{2} \,{\text{and}}\,\sigma_{e}^{2}\) denote the variance components of genotypes, genotype-by-environment interactions and the residuals, t is the number of environments and r is the number of replications. Note that for the second model, \(\sigma_{G}^{2}\) and \(\sigma_{G \times E}^{2}\) were replaced by \(\sigma_{V}^{2}\) and \(\sigma_{V\, \times \,E}^{2}\), as we were interested in the heritability for the genotypes only in experiment II. To get the best linear unbiased estimates (BLUEs) of each genotype, we assumed fixed intercept and genotypic effects, whereas all other effects in the model remained random.
Note that we assumed homoscedasticity of residuals and a compound symmetry model for the genotype-by-environment interaction effects. The model was used for estimating heritability and obtaining the BLUEs of genotypes across environments. In the estimation of heritability, we only need the information on the magnitude of genotype-by-environment interaction through the size of the estimated variance components and it is not necessary to further explore the patterns of interaction. The BLUEs were obtained by necessarily assuming fixed genotypic effects and keeping other effects random. In each experiment, we treated the tested environments as a sample from a TPE, thus it is reasonable to assume a common environmental variance. Since the genotypic effect was assumed to be fixed, this naturally leads to a compound symmetry model for GxE effects. On the other hand, it was reported that the difference between models assuming homoscedastic and heteroscedastic residuals was small and the model assuming homoscedastic residuals could provide acceptable results (Möhring and Piepho 2009).
Genotypic data and analyses
Varieties in both data sets were genotyped with a 90k Infinium single nucleotide polymorphism (SNP) array (Wang et al. 2014). Quality control for the SNP markers was performed to exclude those with missing rates above 5%, rates of heterozygotes above 5%, and minor allele frequencies below 5%. In total 17,839 markers remained in our study.
To investigate the genetic relatedness among the genotypes within and across the two data sets, we estimated the Rogers’ distance (Rogers 1972) for each pair of genotypes based on their marker profiles. More precisely, the Rogers’ distance between individuals i and j is \(d_{ij} = \frac{1}{2m}\mathop \sum \nolimits_{k = 1}^{m} |x_{ik} - x_{jk} |\), where m is the number of markers, \(x_{ik} \,{\text{and}}\,x_{jk}\) are the profiles of the kth marker (being 0, 1 or 2) for individuals i and j, respectively. Distributions of pairwise Rogers’ distances for all genotypes within and across the two data sets were compared. We also performed principal coordinate analysis (PCoA) based on the matrix of Rogers’ distances.
Association mapping and MAS
A two-step association mapping approach was applied in this study (Jiang et al. 2015). First, the BLUE for each genotype in each environment was estimated in a linear mixed model assuming fixed genotype and random replication effects. Then a standard linear mixed model (Yu et al. 2006) was implemented for a genome-wide association mapping scan:
where y il is the BLUE for the ith genotype in the lth environment, μ is a common intercept term, g i is the effect of the ith genotype, E l is the effect of the lth environment, a is the effect of the marker being tested, m is the vector of marker record and e il is the residual term. In the model we assume random genotypic and environmental effects. The effect of the marker being tested was assumed to be fixed. The population structure was considered by assigning a kinship matrix as the variance–covariance matrix for the random genotypic effects. The entries in the kinship matrix were 1 minus the Rogers’ distances. The environmental effects and the residuals were assumed to be independently normally distributed. Parameters were estimated by a restricted maximum likelihood (REML) approach. Significance of marker effect was tested based on the Wald statistic.
Note that we combined the BLUEs obtained in each environment as the response variable. It is expected that this model can increase power of association mapping, compared with the model directly using BLUEs across environments (Stich et al. 2008).
We applied three different significance thresholds (P < 0.005, 0.001, and 0.0001) to study their influence on the accuracy of MAS. For each threshold, significant markers were determined and then fitted together in a multiple linear regression model to obtain the estimation of their effects. Instead of directly recording the number of significant markers, we estimated the effective number of marker–trait associations to account for possible linkage disequilibrium (Jiang et al. 2015). For this purpose, we first performed principal component analysis with all significant markers, and then extracted the minimal number of principal components needed to portray 90% of the total variation. This number approximated the number of independent genetic factors underlying FHB resistance. Note that the threshold of percentage can be chosen arbitrarily, while we chose 90% in order to get results comparable with our previous study (Jiang et al. 2015).
Genomic predictions
We used three different models for GS: ridge regression best linear unbiased prediction (RR-BLUP; Meuwissen et al. 2001; Whittaker et al. 2000), reproducing kernel Hilbert space regression (RKHSR; Gianola and van Kaam 2008), and Bayes-Cπ (Habier et al. 2011). Among the three models, RR-BLUP and Bayes-Cπ exploited the additive effects of the markers across the genome, while RKHSR implicitly modeled additive-by-additive epistatic effects among the markers (Jiang and Reif 2015). We briefly described the three models as follows.
Let n be the number of genotypes, p be the number of markers and l be the number of environments. Let X = (x ij ) be the n × p matrix of markers with x ij being the number of a chosen allele at the jth locus for the ith genotype. Let y be the n-dimensional vector of phenotypic records, which are BLUE of genotypic values obtained in the phenotypic data analyses. Let 1 n be the n-dimensional vector of 1s. In the following models, μ always denotes the common intercept term and e denotes the residual term.
The RR-BLUP model has the form \(y = 1_{n} \,\mu + X\alpha + e\), where α is the vector of additive effects of markers. In the model we assume that \(\alpha \sim N\left( {0,I\sigma_{\alpha }^{2} } \right)\), \(e \sim N(0, I\sigma_{e}^{2} )\), where I is the identity matrix, \(\sigma_{\alpha }^{2} = \sigma_{G}^{2} /p\) and \(\sigma_{e}^{2} = \sigma_{R}^{2} /l\). Note that \(\sigma_{G}^{2}\) and \(\sigma_{R}^{2}\) are the estimated genotypic and residual variances in the phenotypic data analyses. The estimation of α is given by the mixed model equations (Henderson 1975).
The Bayes-Cπ model has the same basic setting \(y = 1_{n} \,\mu + X\alpha + e\) as RR-BLUP but with different assumptions. Let \(\alpha_{j}\) be the jth entry of α (\(j = 1, \ldots , p\)). Then α j is assumed to be zero with probability π and \(\alpha_{j} \sim N\left( {0,\sigma_{\alpha }^{2} } \right)\) with probability (1 − π), where π is a random variable whose prior distribution is uniform on the interval [0,1]. The variance component \(\sigma_{\alpha }^{2}\) has a scaled inverse Chi-squared prior distribution with degree of freedom v α and scale \(S_{\alpha }^{2}\). The prior distribution of the residual is \(e \sim N\left( {0, I\sigma_{e}^{2} } \right)\) and \(\sigma_{e}^{2}\) also has a scaled inverse Chi-squared prior distribution with degree of freedom v e and scale \(S_{e}^{2}\). Parameters v α and v e were both set to be 4. \(S_{e}^{2}\) and \(S_{\alpha }^{2}\) are derived following Habier et al. (2011). A Gibbs sampler algorithm was implemented to infer the parameters in the model which was run for 10,000 iterations with a burn-in of the first 1000 iterations.
The RKHSR model is of the form \(y = 1_{n} \,\mu + K\alpha + e\), where \(y\), \(1_{n} \,\mu\) and e are the same as in the RR-BLUP model, \(\alpha \sim N(0,K^{ - 1} \sigma_{\alpha }^{2} )\) is a vector of random effects and K is the n × n symmetric positive-definite matrix whose entries are defined by \(K_{ij} = \exp \left[ {\frac{{(x_{i} - x_{j} )^{'} (x_{i} - x_{j} )}}{h}} \right]\), where x i and x j are (m × 1) vectors of marker indices for the ith and jth genotype, respectively, and h is a smoothing parameter. To determine h and estimate \(\sigma_{\alpha }^{2}\), we first chose a grid of values for h. For each value of h we estimated \(\sigma_{\alpha }^{2}\) using a REML approach and then calculated the fitted values of the model. Finally, we chose the value h optimizing the generalized cross-validation (GCV) statistic of the model.
Validation scenarios
For both MAS and GS, we applied three different validation scenarios: cross-validation sampling genotypes (CV-G), cross-validation sampling genotypes and environments (CV-GE) and independent validation (IV).
In the CV-G scenario, only the data from experiment I was involved in the analyses. In each run of cross-validation, 80% of the 372 genotypes were randomly assigned as the training set (297 genotypes) and the remaining 20% formed the validation set (75 genotypes). The BLUEs of genotypes across all environments were used as observed phenotypic records in both MAS and GS, except in association mapping where the BLUEs in single environments were considered. In MAS, we performed association mapping in the training set and recorded the significant markers. We estimated the effects of the significant markers, which were then used to predict the performance of the genotypes in the validation set. In GS, we estimated the effects of all markers using the training set (RR-BLUP and Bayes-Cπ) or exploited the relationship between the genotypes in the training set and the validation set through the marker-derived kernel matrix of RKHSR and then predicted the performance of the genotypes in the validation set. The whole procedure was repeated 100 times.
In the CV-GE scenario, we mimicked the situation that the genotypes in the training and the validation set were tested in different years focusing again on the experiment I. In each run, 80% of the 372 genotypes and two environments with the same year (2009 or 2010) were randomly sampled as the training set. The remaining 20% of the genotypes and the other two environments formed the validation set. MAS and GS models were implemented in the same way as in CV-G, except that the BLUEs across the corresponding two environments for the genotypes in the training and validation sets were used instead of the BLUEs across all four environments. In the CV-GE scenario, only the RR-BLUP model was implemented for GS. The reason is the following: in our previous study (Jiang et al. 2015), we did not detect any large effect QTL in experiment I. Hence RR-BLUP is more appropriate to access prediction accuracy than Bayes-Cπ. Moreover, we observed only marginal difference between the accuracies of RR-BLUP and RKHSR. Hence we decided to use only RR-BLUP in the CV-GE scenario in order to reduce the computational load.
In scenario IV, both data sets were involved. In MAS, we performed association mapping using the full data set from experiment I as the training set (372 genotypes) and used the identified significant markers to predict for experiment II as the test set (133 genotypes). In GS, the effects of all markers were estimated using the training set (RR-BLUP and Bayes-Cπ) or the marker-derived kernel matrix was estimated (RKHSR) and then we predicted the performance of the genotypes in the test set.
For both MAS and GS, the prediction ability was defined as the Pearson product-moment correlation between predicted and observed genotypic values in the test set (IV) or validation set (CV-G and CV-GE). The prediction accuracy was defined as the prediction ability divided by the square root of the corresponding heritability. In the CV-G and CV-GE scenarios, the mean prediction ability or accuracy was taken across 100 cross-validation runs.
Reliability and prediction accuracy
We considered the genomic best linear unbiased prediction (GBLUP; VanRaden 2008) model: \(y = \mu + Zg + e\), where y, μ and e are as before, g is the vector of genotypic values, Z is the corresponding design matrix. We assumed that \(g \sim N(0,G\sigma_{g}^{2} )\), where G is the genomic relationship matrix (VanRaden 2008), and \(e \sim N(0, I\sigma_{e}^{2} )\). The reliability of the estimated genotypic value of the ith genotype was defined as the correlation between the true and estimated values: \(r_{i} = cor(g_{i} ,\hat{g}_{i} )\). This metric can be calculated as \(r_{i} = \sqrt {1 - \frac{{var(g_{i} ,\hat{g}_{i} )}}{{\sigma_{g}^{2} }}}\), where \(var(g_{i} - \hat{g}_{i} )\) is the squared standard error or the prediction error variance (PEV) of \(\hat{g}_{i}\) (Henderson 1975). Note that the reliability is a statistical parameter measuring the prediction accuracy of each individual.
We applied the GBLUP model in scenario IV. So in the model y is the BLUEs across environments for the 372 genotypes in the training set, g is the vector of genotypic values for all 505 varieties in the training and the test set. In this way, we obtained the reliabilities of the 133 genotypes in the test set. Note that we only need the phenotypic data of the training set and the genotypic data of both sets to estimate the reliabilities of genotypes in the test set. So the difference of the year interaction effects between the two data sets would not be reflected in the estimation. To investigate the predictability of genotypes with different reliabilities, we divided the test set into four subsets according to the reliabilities of the genotypes and then compared the prediction accuracies of MAS and GS across subsets.
In this study, all statistical models were implemented using R (R Core Team 2015) and Asreml-R (Gilmour et al. 2009).
Results
Intensive field evaluations coupled with artificial inoculation resulted in high-quality phenotypic data
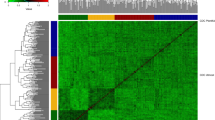
Two panels of European wheat varieties have been assessed for FHB resistance in multi-environmental field trials using artificial inoculations. We observed a broad range of BLUEs resulting in estimates of heritability of 0.91 in experiment I and of 0.74 in experiment II (Table 1). In total, 18 varieties were tested in both experiments. These overlapping genotypes facilitated a combined analysis across both data sets, which revealed that genotype-by-year interaction effects contributed only 7% of the total phenotypic variance. This is also reflected when inspecting the pattern of pairwise correlation coefficients of BLUEs at single environments using 18 common genotypes across the two experiments with no clustering of environments according to their years (Fig. 1a). Considering that the number of common genotypes was small, we also estimated the correlation of BLUEs at single environments for each experiment separately (Fig. 1b, c). For each pair of environments tested in the same experiment, the correlation estimated using all genotypes slightly decreased, compared with the value estimated using the 18 common genotypes. However, the mean difference was only 0.11. In summary, the intensive field evaluation resulted in high-quality phenotypic data representing an excellent source for studying the potential of MAS and GS for FHB resistance.

Heat map of the correlation coefficients among single-environment BLUEs. a The correlations were calculated based on 18 common genotypes evaluated across all seven environments involved in the two experiments. b The correlations were calculated based on 372 genotypes evaluated across four environments in experiment I. c The correlations were calculated based on 133 genotypes evaluated across three environments in experiment II. An environment is a combination of location and year. The abbreviations of the locations are as follows: AHL Ahlum, BOD Halle-Bodenwerde, CEC Cecilienkoog, HEY Heyen, HUZ Hunzen
Genetic diversity within the two data sets is comparable to genetic diversity between them
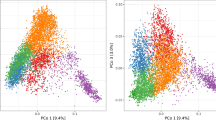
Experiment I comprised 372 European wheat varieties released between 1975 and 2009. In contrast, experiment II included varieties released after 2007. Despite this difference in the year of release, we observed that the Rogers’ distances within the two sets of genotypes did not differ from the Rogers’ distances between lines of the two sets (Fig. 2). This comparable diversity within and between the two sets is further supported by the PCoA of all 505 wheat varieties, which revealed absence of a major genetic differentiation between the two populations (Fig. 3).

Distributions of pairwise Rogers’ distances for all genotypes in experiment I (372 varieties), experiment II (133 varieties) and across two sets

Principal coordinate analysis (PCoA) of the 505 lines (372 lines in experiment I and 133 lines in experiment II) based on Rogers’ distances. Percentages in brackets refer to the proportion of variance explained by the principal coordinate
Prediction accuracies of MAS
We observed in all validation scenarios that the prediction accuracies increased when relaxing the significance threshold from P < 0.0001 to P < 0.005 (Table 2). In scenario IV, the −log10(P) values for all markers were shown in Supplementary Fig. S1. Prediction accuracies of MAS validated in independent samples were for all applied significance thresholds substantially lower than those estimated through cross-validation accounting for genotype sampling with an average difference of 0.19. The relaxed significance threshold resulted in an up to 20-fold increase in the number of putative quantitative trait loci (QTL). Interestingly, prediction accuracies validated in an independent sample were also lower compared to cross-validation accounting for genotype and environmental sampling with an average difference of 0.17 (Fig. 4).

Distribution of prediction abilities for marker-assisted and genomic selection (MAS and GS, respectively) in the fivefold cross-validation scenario sampling genotypes and environments (CV-GE) based on the 372 varieties evaluated in four environments (experiment I). In each fold, 80% of the 372 varieties were considered as training set and phenotypic data from only two environments were used to train MAS (MAS_0.0001, MAS_0.001, and MAS_0.005 at P values <0.0001, 0.001, and 0.005, respectively) and GS. The remaining 20% formed validation set and the BLUEs across the other two environments were used as observed values. The three horizontal lines show the mean prediction abilities observed by only sampling genotypes within the training set (CV-G), along with those obtained by CV-GE and independent validation (IV) scenarios
Prediction accuracies of GS
We examined the prediction accuracies for three GS models using validations in an independent sample as well as applying fivefold cross-validation (Table 2). The prediction accuracies of the three GS models varied slightly with a maximum difference of 8%. Prediction accuracies validated on an independent sample were lower than those estimated through cross-validation accounting for genotype sampling. Nevertheless, prediction accuracies validated on an independent sample were comparable with those from cross-validation accounting for genotype and environmental sampling with an average difference of 0.02 (Fig. 4).
Prediction ability for individual genotypes
The reliability criterion is purely based on the genomic profiles of the lines and has been applied to estimate the prediction accuracy of individual genotypes in the context of GS. The reliability values of the individuals in the test set showed a broad variation from 0.49 to 0.91 (Fig. 5a). We subdivided the validation population into 4 subsets, in which the estimated reliability of genotypes fell into four different ranges (<0.65, 0.65–0.73, 0.73–0.83 and >0.83). We observed an increase in prediction abilities for GS from 0.24 for individuals with reliability values <0.65 to 0.83 for individuals with reliability values >0.8 (Fig. 5b). This trend was also observed for MAS albeit with a lower average level of prediction abilities (Fig. 5b).

a Distribution of reliabilities of the estimated Fusarium head blight resistance values in the genomic best linear unbiased prediction model for the 133 wheat varieties in the test set. b Genomic selection (GS) and marker-assisted selection (MAS) prediction abilities of the 133 wheat varieties in the test set subdivided into four groups according to their reliabilities. Standard errors of the estimated prediction abilities are indicated by segments. The corresponding number of genotypes in each group was indicated in the bracket in the main figure. Three different thresholds (P < 0.005, 0.001, and 0.0001) were used to identify significant markers for MAS
Discussion
Reduced temporal dynamics in allele frequencies with ongoing wheat breeding
Previous diversity studies based on European winter wheat lines reported a major genetic bottleneck which occurred during the green revolution (Boeven et al. 2016; Huang et al. 2007; Roussel et al. 2005). Breeding efforts after the green revolution have caused also systematic shifts in allele frequencies in European winter populations albeit much less pronounced (Boeven et al. 2016; Huang et al. 2007). In accordance with previous findings, we observed in particular for lines released more recently a slow temporal dynamic in allele frequencies with ongoing breeding (Supplementary Fig. S2). This has to be considered when interpreting the prediction accuracies estimated for MAS and GS, which are both driven by relatedness between the training and test populations (Gowda et al. 2014; Habier et al. 2007).
Cross-validated prediction accuracies of MAS and GS evaluated by sampling genotypes are similar to those reported previously
The trends in prediction accuracies observed for MAS for FHB resistance in wheat (Table 2) are in accordance with earlier findings (Jiang et al. 2015; Mirdita et al. 2015a): accuracies increased with relaxed significance thresholds and amounted to ~0.6 when using ~30 independent loci for MAS. The surprisingly high prediction accuracies of MAS for the complex trait FHB resistance can be explained by the exploitation of relatedness between the training and validation sets as discussed in detail by Gowda et al. (2014).
We observed for the three GS approaches prediction accuracies for FHB resistance of around 0.7 (Table 2), which is similar to results reported for panels of European (Jiang et al. 2015; Mirdita et al. 2015a, b) or US wheat lines (Arruda et al. 2015; Rutkoski et al. 2012). In summary, the experimental data underlying our study are representative being an interesting nucleus to contrast prediction accuracies estimated through cross-validation versus validation with independent samples.
Prediction accuracies of GS determined in independent samples are similar to those estimated with cross-validation sampling genotype and environments
The predicting accuracy of GS most often has been estimated performing cross-validation sampling genotypes but not environments (e.g., Crossa et al. 2010; Hofheinz et al. 2012; Iwata and Jannink 2011; Jan et al. 2016). Nevertheless, this leads to an overestimation of the potential of GS which resulted in our study in up to 12% inflated estimates of prediction accuracies (Table 2). The inflation was not severely impacted by population differentiation between the training set and the test set (Figs. 2, 3). Consequently, unbiased estimates of prediction accuracies can be obtained also by cross-validation sampling genotypes and environments (Fig. 4). Our findings are in accordance with results for MAS (Utz et al. 2000) and GS (Schulz-Streeck et al. 2013) in biparental populations leading to the recommendation to apply cross-validation sampling genotypes and environments for obtaining reliable estimates of the prediction accuracy. Nevertheless, it is well known from the cross-validation schemes used in the present study that the training set size for the independent validation is larger than that considered for cross-validation with simultaneous sampling of genotypes and environments. Furthermore, the positive relationship between training set size and prediction accuracy is well documented in the literature (Daetwyler et al. 2008; Endelman et al. 2014; Krchov and Bernardo 2015). In order to rule out that the similarities (in terms of accuracies) between both validation methods were actually an artifact caused by the differences in training set sizes, we re-estimated the prediction ability of RR-BLUP for the independent validation scenario by randomly sampling a subset of 297 individuals from the full training set. Then, the average predictability of 100 samples was only 0.01 lower than the predictability obtained by using the full training set. On the other hand, the standard deviation of the predictability in 100 samples was above 0.02. Hence the 0.01 difference between the prediction abilities obtained by using the reduced and the full training set can be considered marginal. As a result, no biases due to the differences in training set sizes are expected in our findings. It is important to note, however, that this holds true only if population differentiation between the training and validation sets is not pronounced. For European wheat breeding, which is so far characterized by a slow temporal dynamic in allele frequencies (Fig. 2; Boeven et al. 2016), this assumption seems to be realistic. One important consequence is that GS models used to improve European wheat populations likely possess a long validity.
Prediction accuracies of MAS are biased if estimated by cross-validation
In contrast to our results for GS, we observed on average a 126.8% upward bias of the estimates of prediction accuracies of MAS by cross-validation sampling genotypes and environments in comparison to validation with an independent sample (Fig. 4). This finding suggests that MAS is more severely impacted by marginal differences between the genetic composition of the training and validation set as compared to GS. Hence, the evaluation of the potential of MAS cannot be precisely approximated by cross-validation sampling genotypes and environments; thus, validation using independent samples is recommended.
Reliability is not only useful for GS, but also for MAS
Results from a simulation study (Clark et al. 2012) showed that reliabilities are closely associated to the maximum level of relatedness between training set and the particular predicted individual. This suggests that highly reliable predictions would be expected for genotypes that were very well represented by a few or even by a single closely related individual(s) in the training set. Recently, a study based on a large European winter wheat population (He et al. 2016) has demonstrated the value of applying the reliability criterion in the context of GS in order to evaluate the prediction accuracy of individual genotypes. We confirmed this finding and observed that the prediction ability of GS is nearly four times larger for individuals with high reliability values above 0.8 (Fig. 5b). Interestingly, the same holds true also for MAS with an up to six times larger prediction ability for individuals with high reliability values above 0.8. This can be explained on one hand by the role of relatedness driving also the prediction accuracy in MAS (Gowda et al. 2014). On the other hand, marker effects are impacted by genetic background effects (Mackay 2009). Thus, QTL information can be exploited more precisely if the genotype to be predicted is more related to the training population. Consequently, despite being developed in the context of GS, the reliability criterion is a valuable tool to judge the level of prediction accuracy of individual genotypes in MAS.
Author contribution statement
YJ, AWS and JCR wrote the manuscript. YJ performed the calculations. EE and JL contributed to the phenotypic data analyses. BR, JP, SK, EE, VK, OA, GS, MSR and MWG contributed plant material and produced the genomic and phenotypic data.
Abbreviations
- BLUE(s):
-
Best linear unbiased estimator(s)
- FHB:
-
Fusarium head blight
- GBLUP:
-
Genomic best linear unbiased prediction
- GS:
-
Genome-wide selection
- MAS:
-
Marker-assisted selection
- PCoA:
-
Principal coordinate analysis
- PEV:
-
Prediction error variance
- QTL:
-
Quantitative trait loci
- REML:
-
Restricted maximum likelihood
- RKHSR:
-
Reproducing kernel Hilbert space regression
- RR-BLUP:
-
Ridge regression best linear unbiased prediction
- SNP:
-
Single nucleotide polymorphism
- TPE:
-
Target population of environments
References
Arruda MP, Brown PJ, Lipka AE, Krill AM, Thurber C, Kolb FL (2015) Genomic selection for predicting head blight resistance in a wheat breeding program. Plant Genome 8:1–12. doi:10.3835/plantgenome2015.01.0003
Atlin GN, Baker RJ, McRae KB, Lu X (2000) Selection response in subdivided target regions. Crop Sci 40:7–13
Boeven PH, Longin CFH, Würschum T (2016) A unified framework for hybrid breeding and the establishment of heterotic groups in wheat. Theor Appl Genet 129:1231–1245
Buerstmayr H, Ban T, Anderson JA (2009) QTL mapping and marker-assisted selection for Fusarium head blight resistance in wheat: a review. Plant Breed 128:1–26. doi:10.1111/j.1439-0523.2008.01550.x
Clark SA, Hickey JM, Daetwyler HD, Van der Werf JHJ (2012) The importance of information on relatives for the prediction of genomic breeding values and implications for the makeup of reference populations in livestock breeding schemes. Genet Sel Evol 44:4. doi:10.1186/1297-9686-44-4
Crossa J, de los Campos G, Pérez P, Gianola D, Burgueño J, Araus JL, Makumbi D, Singh RP, Dreisigacker S, Yan J, Arief V, Banzinger M, Braun HJ (2010) Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 186:713–724
Daetwyler HD, Villanueva B, Woolliams JA (2008) Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS ONE 3:e3395. doi:10.1371/journal.pone.0003395
Endelman JB, Atlin GN, Beyene Y, Semagn K, Zhang X, Sorrells ME, Jannink J-L (2014) Optimal design of preliminary yield trials with genome-wide markers. Crop Sci 54:48–59
Gianola D, van Kaam JB (2008) Reproducing kernel Hilbert spaces regression methods for genomic assisted prediction of quantitative traits. Genetics 178:2289–2303
Gilmour AB, Gogel B, Cullis B, Thompson R (2009) ASReml user guide release 3.0. VSN International, Hemel Hempstead
Gowda M, Zhao Y, Würschum T, Longin CFH, Miedaner T, Ebmeyer E, Schachschneider R, Kazman E, Schacht J, Martinant JP, Mette MF, Reif JC (2014) Relatedness severely impacts accuracy of marker-assisted selection for disease resistance in hybrid wheat. Heredity 112:552–561
Habier D, Fernando RL, Dekkers JCM (2007) The impact of genetic relationship information on genome-assisted breeding values. Genetics 177:2389–2397
Habier D, Fernando R, Kizilkaya K, Garrick D (2011) Extension of the bayesian alphabet for genomic selection. BMC Bioinform 12:186. doi:10.1186/1471-2105-12-186
Hayes BJ, Bowman PJ, Chamberlaina J, Goddard ME (2009) Invited review: genomic selection in dairy cattle: progress and challenges. J Dairy Sci 92:433–443
He S, Schulthess AW, Mirdita V, Zhao Y, Korzun V, Bothe R, Ebmeyer E, Reif JC, Jiang Y (2016) Genomic selection in a commercial winter wheat population. Theor Appl Genet 129:641–651
Heffner EL, Sorrells ME, Jannink JL (2009) Genomic selection for crop improvement. Crop Sci 49:1–12. doi:10.2135/cropsci2008.08.0512
Henderson CR (1973) Sire evaluation and genetic trends. J Anim Sci 1973:10–41
Henderson CR (1975) Best linear unbiased estimation and prediction under a selection model. Biometrics 31:423–447
Hjorth JSU (1994) Computer intensive statistical methods: validation, model selection and bootstrap. Chapman and Hall, London
Hofheinz N, Borchardt D, Weissleder K, Frisch M (2012) Genome-based prediction of test cross performance in two subsequent breeding cycles. Theor Appl Genet 125:1639–1645
Huang XQ, Wolf M, Ganal MW, Orford S, Koebner RMD, Roder MS (2007) Did modern plant breeding lead to genetic erosion in European winter wheat varieties? Crop Sci 47:343–349
Iwata H, Jannink JL (2011) Accuracy of genomic selection prediction in barley breeding programs: a simulation study based on the real single nucleotide polymorphism data of barley breeding lines. Crop Sci 51:1915–1927
Jan HU, Abbadi A, Lucke S, Nichols RA, Snowdon RJ (2016) Genomic prediction of testcross performance in canola (Brassica napus). PLoS ONE 11:e0147769. doi:10.1371/journal.pone.0147769
Jarquín D, Crossa J, Lacaze X, Du Cheyron P, Daucourt J, Lorgeou J, Piraux F, Guerreio L, Pérez P, Calus M, Burgueño J, de los Campos G (2014) A reaction norm model for genomic selection using high-dimensional genomic and environmental data. Theor Appl Genet 127:595–607
Jiang Y, Reif JC (2015) Modeling epistasis in genomic selection. Genetics 201:759–768
Jiang Y, Zhao Y, Rodemann B, Plieske J, Kollers S, Korzun V, Ebmeyer E, Argillier O, Hinze M, Ling J, Röder MS, Ganal MW, Mette MF, Reif JC (2015) Potential and limits to unravel the genetic architecture and predict the variation of Fusarium head blight resistance in European winter wheat (Triticum aestivum L.). Heredity 114:318–326
Kollers S, Rodemann B, Ling J, Korzun V, Ebmeyer E, Argillier O, Hinze M, Plieske J, Kulosa D, Ganal MW, Röder MS (2013) Whole genome association mapping of Fusarium head blight resistance in European winter wheat (Triticum aestivum L.). PLoS ONE 8:e57500. doi:10.1371/journal.pone.0057500
Krchov LM, Bernardo R (2015) Relative efficiency of genomewide selection for testcross performance of doubled haploid lines in a maize breeding program. Crop Sci 55:2091–2099
Lande R, Thompson R (1990) Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 124:743–756
Mackay TFC (2009) Q&A: genetic analysis of quantitative traits. J Biol 8:23. doi:10.1186/jbiol133
Malosetti M, Bustos-Korts D, Boer MP, van Eeuwijk FA (2016) Predicting responses in multiple environments: issues in relation to genotype × environment interactions. Crop Sci 56:2210–2222
Melchinger AE, Utz HF, Schön CC (1998) Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 149:383–403
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Miedaner T, Wilde F, Korzun V, Ebmeyer E, Schmolke M, Hartl L, Schön C (2009) Marker selection for Fusarium head blight resistance based on quantitative trait loci (QTL) from two European sources compared to phenotypic selection in winter wheat. Euphytica 166:219–227
Miedaner T, Würschum T, Maurer HP, Korzun V, Ebmeyer E, Reif JC (2011) Association mapping for Fusarium head blight resistance in European soft winter wheat. Mol Breed 28:647–655
Mirdita V, He S, Zhao Y, Korzun V, Bothe R, Ebmeyer E, Reif JC, Jiang Y (2015a) Potential and limits of whole genome prediction of resistance to Fusarium head blight and Septoria tritici blotch in a vast Central European elite winter wheat population. Theor Appl Genet 128:2471–2481
Mirdita V, Liu G, Zhao Miedaner T, Longin CFH, Gowda M, Mette MF, Reif JC (2015b) Genetic architecture is more complex for resistance to Septoria tritici blotch than to Fusarium head blight in Central European winter wheat. BMC Genomics 16:430. doi:10.1186/s12864-015-1628-8
Möhring J, Piepho HP (2009) Comparison of weighting in two-stage analysis of plant breeding trials. Crop Sci 49:1977–1988
Paul PA, McMullen MP, Hershman DE, Madden LV (2010) Meta-analysis of the effects of triazole-based fungicides on wheat yield and test weight as influenced by Fusarium head blight intensity. Phytopathology 100:160–171
R Core Team (2015) R: A language and environment for statistical computing. R foundation for statistical computing, Vienna. ISBN 3-900051-07-0. http://www.R-project.org
Reynolds MP, Bonnett D, Chapman SC, Furbank RT, Manes Y, Mather DE, Parry MAJ (2011) Raising yield potential of wheat. I. Overview of a consortium approach and breeding strategies. J Exp Bot 62:439–452
Rogers JS (1972) Measures of genetic similarity and genetic distance. Stud Genet 7:145–153
Roussel V, Leisova L, Exbrayat F, Stehno Z, Balfourier F (2005) SSR allelic diversity changes in 480 European bread wheat varieties released from 1840 to 2000. Theor Appl Genet 111:162–170
Rutkoski J, Benson J, Jia Y, Brown-Guedira G, Jannink JL, Sorrells ME (2012) Evaluation of genomic prediction methods for Fusarium head blight resistance in wheat. Plant Genome 5:51–61
Saint Pierre C, Burgueño J, Crossa J, Dávila GF, López PF, Moya ES, Moreno JL, Muela VMH, Villa VMZ, Vikram P, Mathews K, Sansaloni C, Sehgal D, Jarquin D, Wenzl P, Singh S (2016) Genomic prediction models for grain yield of spring bread wheat in diverse agro-ecological zones. Sci Rep 6:27312. doi:10.1038/srep27312
Schulz-Streeck T, Ogutu JO, Gordillo A, Karaman Z, Knaak C, Piepho HP (2013) Genomic selection allowing for marker-by-environment interactions. Plant Breed 132:532–538
Smith AB, Cullis BR, Thompson R (2005) The analysis of crop cultivar breeding and evaluation trials: an overview of current mixed model equations. J Agric Sci 143:449–462
Stich B, Melchinger AE, Heckenberger M, Möhring J, Schechert A, Piepho HP (2008) Association mapping in multiple segregating populations of sugar beet (Beta vulgaris L.). Theor Appl Genet 117:1167–1179
Utz HF, Melchinger AE, Schön CC (2000) Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154:1839–1849
VanRaden P (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
VanRaden P, Van Tassell C, Wiggans G, Sonstegard T, Schnabel R, Taylor J, Schenkel F (2009) Invited review: reliability of genomic predictions for North American Holstein bulls. J Dairy Sci 92:16–24
Wang S, Wong D, Forrest K, Allen A, Chao S, Huang BE, Maccaferri M, Salvi S, Milner S, Cattivelli L, Mastrangelo AM, Whan A, Stephen S, Barker G, Wieseke R, Plieske J, IWGSC, Lillemo M, Mather D, Appels R, Dolferus R, Brown-Guedira G, Korol A, Akhunova A, Feuillet C, Salse J, Morgante M, Pozniak C, Luo M, Dvorak J, Morell M, Dubcovsky J, Ganal M, Tuberosa R, Lawley C, Mikoulitch I, Cavanagh C, Edwards KJ, Hayden M, Akhunov E (2014) Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796
Whittaker JC, Thompson R, Denham MC (2000) Marker-assisted selection using ridge regression. Genet Res 75:249–252
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhao Y, Li Z, Liu G, Jiang Y, Maurer HP, Würschum T, Mock HP, Matros A, Ebmeyer E, Schachschneider R, Kazman E, Schacht J, Gowda M, Longin CFH, Reif JC (2015a) Genome-based establishment of a high-yielding heterotic pattern for hybrid wheat breeding. Proc Nat Acad Sci USA 112:15624–15629. doi:10.1073/pnas.1514547112
Zhao Y, Mette MF, Reif JC (2015b) Genomic selection in hybrid breeding. Plant Breed 134:1–10. doi:10.1111/pbr.12231
Acknowledgements
The phenotypic and genotyping data were generated within the frames of the projects GABI-Wheat and VALID (Project numbers 0315067 and 0315947) funded by the Plant Biotechnology program of the German Federal Ministry of Education and Research (BMBF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SK, EE, VK are employed by the company KWS LOCHOW GMBH. OA, GS are employed by Syngenta Seeds and JP, MWG are employed by the company TraitGenetics GmbH. The companies have commercial interest in the results for application in variety development and for the provision of genotyping services. This does not alter the authors’ adherence to the policies of Theoretical and Applied Genetics on sharing data and materials.
Additional information
Communicated by M. Malosetti.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jiang, Y., Schulthess, A.W., Rodemann, B. et al. Validating the prediction accuracies of marker-assisted and genomic selection of Fusarium head blight resistance in wheat using an independent sample. Theor Appl Genet 130, 471–482 (2017). https://doi.org/10.1007/s00122-016-2827-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-016-2827-7