
Abstract
The use of molecular markers to detect polymorphism at DNA level is one of the most significant developments in molecular biology techniques. With the development of new next-generation sequencing technologies, the discovery of SNP became easier and faster, and the costs of data point were reduced. The development and use of SNP markers for coffee have provided new perspectives for the evaluation of genetic diversity and population structure via different statistical approaches. In this study, 72 Coffea canephora genotypes were analyzed to identify the SNP markers and apply them to genetic studies and selection of parents/hybrids in genetic breeding. As many as 117,450 SNP were identified using the RAPiD Genomics platform. After quality analyses, 33,485 SNP were validated for analyses of genetic diversity and population structure. Genotypes were separated based on their varietal groups, and Hybrids were differentiated using the clustering and Bayesian approach. Coffee accessions mistakenly identified in the germplasm and breeding program were detected. The Conilon varietal group presented the lowest genetic dissimilarity values, suggesting the introduction of new accessions in the germplasm bank. The highest genetic distances values were observed among genotypes of the heterotic groups (Conilon and Robusta). The markers were efficient in evaluating the genetic diversity and population structure of C. canephora. Promising crosses were selected within and between the varietal groups. Hybrids with greater genetic distances were selected, which were important for C. canephora breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The use of molecular markers to detect polymorphism at DNA level is one of the most significant developments in molecular biology techniques. Molecular markers are more advantageous than phenotypic markers for presenting the highest selection gain for the traits of interest and for being uninfluenced by the environment. In addition, they can be used at the plant development stage (Gartner et al. 2013).
Among the several types of molecular markers, SNP have stood out for being the most abundant type of polymorphism in genomes, for being codominant and biallelic, and for automatizing the technique used to obtain these markers (Resende et al. 2008; Liao and Lee 2010). In recent years, these markers have frequently been used in studies on humans (Gabriel et al. 2002; Ojopi et al. 2004; Pinto et al. 2008; Gnirke et al. 2009) and in some animal and plant species (Abatepaulo et al. 2008; Caetano 2009; Yang et al. 2013); (Zhang et al. 2011; Neves et al. 2013, 2014; Zhang et al. 2015; Resende et al. 2016). However, they have not yet been identified in several plant species of economic relevance, such as the coffee species Coffea canephora, as they are relatively recent.
With the development of new next-generation sequencing (NGS) technologies, the discovery of SNP became easier and faster, and the costs of data point were reduced (Carvalho and Silva 2010).
The company RAPiD Genomics, located in Florida, USA, developed a methodology to obtain SNP using NGS developed for humans (Gnirke et al. 2009) and adapted to plants (Neves et al. 2013, 2014). This technology uses a method of genotyping-by-sequencing of specific regions of the genome.
Once obtained, the SNP can be used in several relevant studies of the species, such as in the analyses of genetic diversity and population structure. These analyses validate SNP to evaluate the discriminatory capacity in genetically related populations, besides the genetic variability and population structure of the species studied.
The presence of genetic variability is a basic and necessary condition for success in a genetic breeding program. The genetic variability observed in C. canephora species is mostly due to the large geographic distribution (especially of the genus Coffea), the domestication process, and the natural allogamy observed in the species (Davis et al. 2006; Babova et al. 2016).
The diversity of C. canephora was first described by Berthaud (1986). The author identified two distinct genetic groups based on their respective centers of diversity: the Guinean group, formed by West African genotypes (Guinea and Ivory Coast), and the Congolese group, made up of Central African genotypes. Other studies have also indicated that the division of C. canephora into two groups is strongly related to its geographic isolation and historical events of glaciations that occurred 18,000 years ago (Gomez et al. 2009; Cubry et al. 2013).
The Congolese group, unlike the Guinean group, was disseminated and progressively improved throughout the domestication and breeding processes of C. canephora. The Congolese group was divided into five subgroups, namely SG1, SG2, B, C, and UW (recently discovered and made up of wild accessions from Uganda) (Musoli et al. 2009).
The subgroup SG1 is formed by genotypes occurring from Benin to Gabon, also known as Conilon coffee. SG1 is the subgroup mostly adapted to Brazil and is present in the main varieties developed in the country. The subgroups SG2 (from the Democratic Republic of Congo), B (from the Central African Republic), and C (from Cameroon) consist of genotypes known as Robusta coffee. These coffee plants are tall, vigorous, with large leaves and fruits, are resistant to coffee rust, and more susceptible to drought (Marraccini et al. 2012).
Conilon and Robusta are considered as two heterotic groups with distinct and complementary traits within the C. canephora species. Thus, the expressive genetic variability observed between and within C. canephora population makes this species extremely important in breeding programs that aim at new varieties that benefit from heterosis (Lashermes et al. 2000; Leroy et al. 2005).
Rapid advances in molecular technologies have opened new perspectives for the evaluation of genetic diversity, involving different statistical approaches (Ferrão et al. 2015). A genetic dissimilarity matrix (or similarity) can be constructed by the analysis of all possible pairwise genotypic combinations, which can subsequently be used to establish patterns of similarities and dissimilarities (Kosman and Leonard 2005). A Bayesian approach implemented in the STRUCTURE software (Pritchard et al. 2000) has also been used to study the genetic diversity and population structure. In this software, genotypic data are used for the probabilistic classification of genotypes considering K populations (in which K may be unknown).
Therefore, this study aimed to identify SNP molecular markers for C. canephora species, validate them for genetic studies, and use them to select parents and hybrids for breeding.
Materials and methods
Genetic material
The population was selected to represent a broad genetic variability of the species C. canephora. To this end, divergent materials with agronomic traits of interest were selected.
An interpopulational partial diallel was carried out to form base populations to initiate the recurrent selection program of C. canephora in the breeding program of the Agricultural Research Company of Minas Gerais (Epamig), in association with the Federal University of Viçosa (UFV) and the Brazilian Company of Agricultural Research—Coffee (Embrapa Café). Five genotypes of Conilon (male parent) and five genotypes of Robusta (female parent) were used in the diallel. Besides these parents, five Conilon genotypes, four Robusta genotypes (Table 1), and 20 Hybrids (Conilon × Robusta) obtained from the diallel (Table 2) were also included in the study population. Twenty-one genotypes of the Conilon varietal group and 22 of the Robusta varietal group (Table 3) of the Epamig/UFV/Embrapa breeding program were selected, totaling 72 genotypes.
The genetic material of Conilon was obtained from the Capixaba Institute of Research, Technical Assistance and Rural Extension (INCAPER), and the Robusta material was obtained from the Centro Agronómico Tropical de Investigación y Enseñanza (CATIE). The materials were brought to EPAMIG (Viçosa, MG) germplasm bank by the researcher Antonio Alves Pereira.
Genomic DNA extraction
Young and fully expanded leaves from the 72 genotypes were collected. Genomic DNA was extracted using the methodology described by (Diniz et al. 2005). DNA concentration was verified in NanoDrop 2000, and its quality was evaluated in 1% agarose gel. DNA concentration of the samples was standardized and taken to RAPiD Genomics for the construction of probes, sequencing, and identification of SNP molecular markers.
Probes design and SNP identification
The sequencing-based genotyping was carried out using targeted enrichment, followed by next-generation sequencing, as formerly described (Gnirke et al. 2009; Song et al. 2016). A total of 40,000 120-bp probes were designed, using a combination of genomic resources available for C. arabica and C. canephora, aiming to reduce the complexity of the genome and enrich the libraries for the targeted regions. The genomic resources consisted of C. arabica specific expressed sequence tags (ESTs) and C. canephora specific ESTs obtained from a database of the Brazilian Coffee Genome Project, consisting of more than 200 thousand ESTs and corresponding to about 33 thousand transcripts (Vieira et al. 2006). Moreover, probes from the reference genome of C. canephora (Denoeud et al. 2014) were also developed, with an estimated size of 710 Mb. The EST database was also mapped against the reference genome to determine candidate SNP that represented good candidate regions to become a polymorphic probe. Furthermore, all the probes were designed in regions that were not repetitive in the C. canephora genome and absent in the plastid genome.
The process consisted of targeting ESTs that were unique to C. arabica and did not match the canephora reference genome. Based on these unigenes, 1879 probes derived exclusively from C. arabica contigs were designed. However, the reference genome of C. canephora was published with a total length of about 568.6 Mb, corresponding to 80% of the estimated total length (710 Mb) of the genome this species (Denoeud et al. 2014). Thus, the sequences of these probes were incorporated into the reference genome of this species. Afterward, we identified 8236 probes mapping to 7347 annotated genes that contained at least one SNP between the EST database and the reference genome. Then, the reference genome was used to design an additional set of 11,879 probes that were mapped uniquely in the genome and represented 11,879 genes, besides the 7347 genes formerly described. Finally, to add up to 40,000, 18,006 probes were selected in non-genic regions of the reference genome by breaking the genome in 10 kb intervals and selecting probes that are well-distributed and that covered the entire genome.
The 72 coffee samples were prepared for NGS and hybridized against the probes synthesized in-solution. The analysis used the protocols described by Neves et al. (2013). C. canephora and C. arabica genotypes were sequenced using the sequencing platform Illumina Hi-Seq after capturing 40,000 target regions in the coffee genome. The sequencing product was separated into each individual barcodes, corresponding to each genotyped sample. Low-quality bases with less than 20 quality score in the 3′ end were trimmed out, followed by a low-quality filter that removed reads with more than 10% of the read with less than 20 quality score. Filtered reads were aligned against the reference genome of C. canephora using Mosaik (Lee et al. 2014). Single Nucleotide Polymorphisms (SNP) were identified using diploid settings on Freebayes (Garrison and Marth 2012), leading to the identification of 162,026 SNPs (SNP specific to C. arabica, specific to C. canephora, and common to both species) in 27,651 polymorphic probes, with a mean of five SNP per probe.
Quality analysis of SNP
The VCFtools software (Danecek et al. 2011) was used for quality analysis of the SNPs (Resende et al. 2016). The following quality parameters were used: MinDP—defined as the minimum sequencing depth per SNP and per individual, in which SNP with values lower than that determined in the filter were considered as missing data; DPrange—mean sequencing depth range of the population for which an SNP is maintained. SNP markers with mean coverage in the population outside the range determined in each filter were removed; Miss—percentage of missing data accepted in the population after filtering by the MinDP parameter. SNP markers with Miss values above that set in the parameter were removed; MAF—minor allele frequency that refers to the frequency at which the alternative allele occurs in a given population; and MinQ—minimum quality accepted for SNP. SNP with quality values lower than those established by this criterion were removed.
Two data filtering analyses were applied. To obtain the dataset of the first filter, the parameters MinDP3, DPrange15-750, Miss0.4, MAF0.01, and MinQ10 were considered. For the second filtering, the parameters MinDP5, DPrange20-350, Miss0.2, MAF0.03, and MinQ20 were considered. The graphical analysis was performed using the Circos (Krzywinski et al. 2009).
Analysis of genetic diversity and population structure
The SNP data were coded as 0, 1, and 2 for being biallelic and codominant markers. Thus, considering the genotypes of the individuals as A1A1, A1A2, or A2A2 in each locus, these genotypes were coded as 0, 1, or 2, respectively.
The genetic dissimilarity (or similarity) matrix was obtained by the arithmetic complement of the unweighted index using the GENES software (Cruz 2013). Genetic distance can be estimated by the following equation:
where: Dii′ = genetic distance between pairs of accessions i and i′;
\(p_{j} = \frac{{a_{j} }}{A}\): weight associated with loco j, determined by: aj: total number of loco alleles j; A: total number of alleles studied; cj: number of common alleles between pairs of accessions i and i′.
The cluster analysis to construct the dendrograms was performed using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) and Neighbor-Joining (NJ), in the statistical software MEGA 7.0 (Kumar et al. 2016).
Another cluster analysis was performed using the STRUCTURE software (Pritchard et al. 2000), which considered the K values ranging from 1 to 6, with ten replicates. Each run was carried out with a 25,000 burn-in period, followed by 75,000 MCMC (Markov Chain Monte Carlo). The best number of genetic groups was estimated by calculating the ΔK, as described by Evanno et al. (2005), using the software STRUCTURE HARVESTER (Earl and VonHoldt 2012).
Principal Components Analysis (PCA) was performed using the pcaMethods R package (Stacklies et al. 2007) implemented in ClustVis (Metsalu and Vilo 2015).
Results
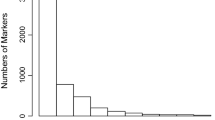
The median alignment was 95% and a total of 117,450 SNP markers for C. canephora were identified with the probes distributed throughout the coffee genome. After the quality analyses, 57,529 SNP markers were obtained with the first filter, and 33,485 SNP markers were obtained with the second filter (Fig. 1). For the genetic analyses, the 33,485 SNPs with the strictest quality parameters were considered in the study. These 33,485 SNP, as well as their respective positions in the genome, were made available (Online Resource 1). The number of SNP per chromosome, considering the second filter, ranged from 124 to 5076. The highest number of SNP was observed on chromosomes 0 and 2 (Fig. 1).

Number of SNP markers by chromosomes obtained from raw data and filters 1 and 2. SNP markers distributed throughout the UNIGENES from the EST sequences of Coffea arabica and from the 11 Coffea canephora chromosomes and chromosome 0 of Coffea canephora
A summary of the distribution of SNP in the genome of C. canephora is presented in layers (Fig. 2). The first (black, from outer to inner layers) represents each chromosome. The second layer corresponds to the number of raw SNP (blue). The last two layers (red and green) express the density of SNP after the first and second filters, respectively.

Circular visualization of the SNP distribution in the Coffea canephora genome. From the outer to the inner layers, the graphic is separated into four layers: (i) Chromosomes; (ii) number of raw SNP; (iii) number of SNP after first filter; (iv) number of SNP after second filter
The genetic distance matrix was generated using the molecular data, and two large groups (I and II) were formed with the dendrogram using the UPGMA technique. The first group was formed by accessions of the varietal group Robusta, and the second group consisted of accessions of the varietal group Conilon and the Hybrids (Conilon × Robusta). The second group can be divided into two subgroups, II. a and II. b, consisting of the accessions of Conilon and Hybrids, respectively (Fig. 3).

Dendrogram obtained using the UPGMA technique, based on the arithmetic complement of the unweighted index of 72 Coffea canephora genotypes (26 Conilons, 26 Robustas, and 20 Hybrids) and 33,485 SNP markers. Green, red, and blue lines represent the varietal groups (Conilon and Robusta) and the Hybrids, respectively
The Hybrid H0915-1 was allocated in the group of Robusta accessions. The genotypes Conilon UFV 3628-2 and UFV 3628-45, together with the genotype Robusta UFV 3374-29 were allocated in the subgroup of Hybrids (Fig. 3).
Results of the NJ algorithm for cluster analysis were equivalent to those obtained by the UPGMA methodology. The two dendrograms allocated the genotypes in the Conilon, Robusta, and Hybrid groups, respectively. The Hybrid H0915-1 was also allocated in the group of Robusta genotypes. The genotypes Conilon UFV 3628-2 and UFV 3628-45 and the genotype Robusta UFV 3374-29 were allocated closer to the subgroup of the Hybrids (Fig. 4).

Neighbor Joining (NJ) dendrogram of the 72 C. canephora genotypes (26 Conilons, 26 Robustas, and 20 Hybrids). Green, red, and blue lines represent the varietal groups (Conilon and Robusta) and the Hybrids, respectively
The Bayesian approach, performed by the STRUCTURE software (Pritchard et al. 2000), confirmed the results of the clustering methods. According to the methodology proposed by Evanno et al. (2005), the highest value of ΔK was obtained for K = 2 (Fig. 5), showing that genotypes can be separated into two groups, based on the Conilon or Robusta varietal group (Fig. 6a).

Graphic obtained with the values of ΔK for visualization of the best K (K = 2), according to the methodology proposed by Evanno et al. (2005)

Bar graphic of the STRUCTURE software used to study the diversity of the 72 Coffea canephora genotypes (26 Conilons, 26 Robustas, and 20 Hybrids). The 72 genotypes are represented below the graphic, and were divided into two (K = 2) and three (K = 3) groups, Fig. 6a, b respectively, according to the Conilon and Robusta varietal group, and also allowed discriminating Hybrid genotypes resulting from crosses between the two heterotic groups. The red bar is associated to the Robusta genotypes, the green bar to the Conilon genotypes and the blue bar to the Hybrid genotypes
The Hybrid H0915-1 presents approximately 80% genetic similarity with Robusta genotypes. The genotypes Conilon UFV 3628-2 and UFV 3628-45 showed approximately 50% genetic similarity with the Conilon group and 50% genetic similarity with the Robusta group. The genotype Robusta UFV 3374-29 showed approximately 80% genetic similarity with the Robusta group and 20% genetic similarity with the Conilon group (Fig. 6a).
Considering the division of the population into three groups (K = 3) (Fig. 6b), Robusta genotypes were allocated to group I (red bars). Group II (blue bars) was composed of the Hybrids and the genotypes Conilon UFV 3628-2 and UFV 3628-45 and the genotypes Robusta UFV 3368-58 and UFV 3374-29. Group III (green bars) clustered the Conilon genotypes. Hybrid H0915-1 was allocated to group II but presented about 38% of genetic mixture with group I. The genotype Robusta UFV 3368-58 was allocated to group II but presented about 42% of genetic mixture with group I. The genotype Robusta UFV 3374-29 was allocated to group II but presented about 31% of genetic mixture with group I and 5% of genetic mixture with group III.
Values of genetic dissimilarities estimated between and within the varietal groups (Conilon and Robusta) and the Hybrids are presented in Tables 4 and 5. Table 4 shows the lowest genetic dissimilarities and the mean distance observed between and within the groups. Table 5 shows the five major genetic dissimilarities obtained between and within the varietal groups and the Hybrids.
The lowest estimates of genetic distances were verified among the genotypes of the Conilon varietal group, and the lowest distance (0.0071) was observed between the accessions Conilon UFV 3628-45 and UFV 3628-2 (Table 4). These genotypes were grouped in the Hybrids subgroup, based on the analyses of the dendrograms.
The highest values of genetic distances were detected between the genotypes of the Conilon and Robusta varietal groups. The highest distance (0.3923) was obtained between the genotypes Conilon UFV 3629-11 and Robusta UFV 3371-19 (Table 5). The lowest genetic distance was obtained between the genotypes Conilon UFV 3628-45 and Robusta UFV 3374-29. According to the analyses of the dendrograms, the two genotypes were allocated to the Hybrid subgroup. The overall mean genetic distance was 0.2395.
The Hybrids H0915-1, H0919-2, and H0921-1 presented the highest values of genetic dissimilarity when compared with other Hybrids and the genotypes of the Conilon and Robusta groups. Among these three Hybrids, the greatest genetic distance was observed between H0915-1 and H0921-1 (0.2284) (Table 5). According to the analyses of the dendrograms, Hybrid H0915-1 was allocated to the Robusta group.
Table 6 shows the five largest distances between and within the varietal groups and Hybrids, disregarding the genotypes Conilon UFV 3628-2 and UFV 3628-45, the genotype Robusta UFV 3374-29, and the Hybrid H0915-1 (for having been mistakenly allocated in their respective groups).
Based on Table 6, genotypes of the Conilon and Robusta varietal groups with greater genetic dissimilarities can be selected to form base populations. Hybrids can also be selected; for instance, Hybrids H0911-2, H0919-2, H0922-1, H0912-1, H0921-1, and H0918-1 stood out for their higher genetic distance values. These Hybrids can be used to compose a set of hybrid varieties and thus prevent problems with genetic autoincompatibility present in C. canephora species.
A PCA was performed using SNP markers, revealing two principal components (Fig. 7). PC1 and PC2 contributed with variations of 68.4 and 19.0%, respectively. Cumulatively, these two PCs contributed with 87.4% of the total variation observed. PCA formed three groups. The genotypes Conilon UFV 3628-45, UFV 3628-2, and UFV 3629-11; the genotype Robusta UFV 3368-58; and the Hybrid H0915-1 were not allocated to any group. The genotype Robusta UFV 3374-29 was allocated to the Hybrids group. The genotypes Conilon UFV 3628-45 and UFV 3628-2, although not allocated to any group, were closer to the Hybrid group. Hybrid H0915-1 was close to the Robusta group. Although genotype Robusta UFV 3368-58 was allocated in any group, it was located between the Robusta and Hybrid groups, corroborating the results of the software STRUCTURE for K = 3 (Fig. 6b).

Analysis of Principal Components of the 72 genotypes of Coffea canephora, showing the formations of three groups. X and Y axis show principal component 1 and principal component 2 that explain 68.4 and 19% of the total variance, respectively
PCA results agree with those obtained in the previous analyses, except for genotype Conilon UFV 3629-11. Although this genotype was not allocated in any group, it was located close to the Conilon group by the PCA analysis. Results also show that Robusta genotypes have greater spatial dispersion (genetic variability) in relation to the Conilon genotypes.
Discussion
Due to the large number of data obtained from genotyping with thousands of molecular markers, even a low error rate can be detrimental in subsequent analyses. Thus, some parameters for quality control and data filtering must be used to remove these errors. These evaluations allow identifying below-standard markers and samples, which must be removed before the analysis to reduce false positive error (type I error) and false negative error (type II error) (Anderson et al. 2010).
The RAPiD Genomics platform presented a large potential for SNP identification and genotyping since a significant number of these markers was identified in C. canephora when compared with other studies (Zhou et al. 2016; Garavito et al. 2016). A greater number of SNP was identified on chromosomes 0 and 2. However, chromosome 0 is not a true chromosome, but a set of unsorted sequence scaffolds of C. canephora (Denoeud et al. 2014). A study using a mixed model to multiple harvest-location trials applied to genomic prediction in Coffea canephora identified the highest number of SNP on chromosome 2 after filtering the data (Ferrão et al. 2017). These results may be due to the length of chromosome 2 in the genome of C. canephora (Denoeud et al. 2014).
These markers were able to divide the genotypes of the studied population into distinct groups. Thus, the efficiency of these SNP markers has been proven in the analyses of genetic diversity and population structure.
A technique based on the reduction of complexity by using the restriction enzymes that aim at gene-rich regions and by using the NGS technology, known as DArTseq, was applied to test the performance of the markers derived from this method in coffee plants. By studying the genetic diversity of the species C. canephora cultivated in Vietnam and Mexico, 4,021 SNPs were identified (Garavito et al. 2016). These SNP markers constitute a valuable tool for breeders. In addition, they increase the knowledge on the genetic diversity of C. canephora and contribute to the understanding of the genetic background of important varieties from major coffee producers (Garavito et al. 2016).
In another study, a set of SNP markers was developed and used to assist the identification of coffee germplasm, demonstrating the usefulness of EST sequences as an approach for the rapid development of a high-throughput targeted genotyping tool (Zhou et al. 2016). By using ESTs of the C. arabica, C. canephora, and C. racemosa obtained from public databases, 7538 SNP markers were developed, of which 180 were selected for validation using 25 accessions of C. arabica and C. canephora from Puerto Rico. Based on the validation result, a panel of 55 SNP markers that are polymorphic between both coffee species was constructed (Zhou et al. 2016).
In the UPGMA, the SNP were able to classify the genotypes into different groups, based on the varietal group (Conilon and Robusta). It also discriminated the Hybrid genotypes obtained from the cross between Conilon and Robusta genotypes. The results were confirmed by the analysis using the NJ algorithm; by the Bayesian approach, using the STRUCTURE software (Pritchard et al. 2000); and by the PCA analysis. These three analyses were also efficient in separating the Conilon and Robusta varietal groups. However, as in the previous UPGMA cluster analysis, the Hybrid genotypes were also discriminated.
Robusta and Conilon heterotic groups present distinct and striking phenotypic traits. Nevertheless, the classification of these materials is no easy task. This fact is due to their strictly allogamous reproduction form, high phenotypic amplitude, and heterozygosis, in addition to possible natural crosses between the two varietal groups. Thus, studies on the genetic diversity using molecular markers are crucial in breeding programs for the conservation of genetic sources and the correct evaluation of genotypes (Ferrão et al. 2013; Ren et al. 2013; Ferrão et al. 2015).
Three accessions (Conilon UFV 3628-2, Conilon UFV 3628-45, and Robusta UFV 3374-29) were not allocated to their respective varietal groups, but to the subgroup of the Hybrids. This result suggests that these accessions probably derived from natural crossings and were mistakenly classified.
Conilon genotypes, including UFV 3628-2 and UFV 3628-45, are derived from half-sib seeds. Since Conilon is allogamous and has an autoincompatibility system, the two seeds that originated these two genotypes may have been originated from the pollination with a Robusta parent, instead of Conilon, forming natural Hybrids in their collection site.
The genotype Conilon UFV 3628-2 was chosen as the parent to obtain Hybrids in the interpopulational partial diallel due to its prominence in yield, resistance to rust, and earliness. The genotype Conilon UFV 3628-45 also stood out for these traits. Resistance to rust is evidence that these accessions may be natural Hybrids since Conilon genotypes are usually susceptible to rust. Resistance to this disease is usually obtained from Robusta (Cubry et al. 2008; Musoli et al. 2009). In addition, relatively high yield was observed in these accessions when compared with the other Conilon genotypes. In the mean of 5 years of evaluations (2012–2016) (data not shown), the genotypes Conilon UFV 3628-2 and Conilon UFV 3628-45 were in the fifth and eighth position, respectively, in yield data, when compared with the other Conilon genotypes evaluated in the breeding program. Yield increase may also be the result of Hybrid vigor, corroborating the molecular data that indicate these natural Hybrid genotypes.
The genotype Robusta UFV 3368-58 probably derived from natural crossings and was also mistakenly classified. When using the software STRUCTURE for K = 3, this genotype was allocated to group II; by the NJ algorithm, it is also located close to the Hybrid genotypes; and in the PCA analysis, despite not being allocated to any group, this genotype is located between the Robusta and Hybrid groups.
The accession Robusta UFV 3374-29, also indicated as a probable natural Hybrid by the molecular data, stood out for the mean yield of the 5 years of evaluations, although data were slightly inferior when compared with those of the accessions Conilon UFV 3628-2 and Conilon UFV 3628-45. This accession is prominent for its earliness, which was probably obtained from its possible parent Conilon. The analysis using the STRUCTURE software for K = 2 revealed approximately 78% similarity with Robusta and approximately 22% similarity with Conilon. This contribution of the Conilon parent appears to be sufficient to guarantee the earliness of the genotype Robusta UFV 3374-29. In the analysis using the software STRUCTURE for K = 3, the genotype Robusta UFV 3374-29 was allocated to group II, proving that this genotype is a natural Hybrids.
The Hybrid H0915-1 was allocated to the Robusta group in both cluster analyses. Based on the molecular data, one of the parents of Hybrid H0915-1 is the genotype Conilon 3628-2, which is a natural Hybrid. Thus, this Hybrid was formed by the cross [(Robusta × Conilon) × Robusta]. This explains why the Hybrid H0915-1 was genetically closer to Robusta genotypes in the cluster analyses. This result was confirmed by the STRUCTURE software for K = 2, which shows approximately 84% similarity with Robusta and approximately 16% similarity with Conilon. Using the STRUCTURE software for K = 3, Hybrid H0915-1 was allocated to group II but presented about 38% of genetic mixture with group I, which comprises Robusta genotypes.
The genetic distance matrix showed that the Conilon varietal group presented the lowest values of genetic dissimilarity. The presence of genetic variability within the base population is responsible for the success of the breeding programs (Grandillo 2014). Thus, to obtain the base population of genotypes of the Conilon varietal group, genotypes with the highest values of genetic distance observed in the distance matrix must be selected. Therefore, in this germplasm, the five crosses presented in Table 6 should be prioritized, if the objective is to obtain crosses only between accessions of the Conilon group. The possibility of increasing the variability of this group in the Germplasm Bank, by introducing new accessions, should also be considered.
To form the base population of the Robusta varietal group, the accessions presented in Table 6 must also be prioritized, since they have the highest values of genetic dissimilarity and are important sources of variability for breeding programs.
Higher genetic distance between genotypes of the Conilon and Robusta heterotic groups were expected. Therefore, the higher distance value (0.3923) between the genotype Conilon UFV 3629-11 and the genotype Robusta UFV 3371-19 is explained by the fact that these genotypes present distinct and complementary traits.
In breeding programs, such as that of Epamig/UFV/Embrapa, which aim at identifying heterotic groups and contrasting parents to explore Hybrid vigor, the correct evaluation of diversity is a key factor for the choice of parents. The expectation that divergent or contrasting parents generate good Hybrids is because if two parents are genetically close, there is a trend for them to share many common genes or alleles. Thus, while crossing them, little complementarity and low Hybrid vigor will occur due to the low level of allelic heterozygosity in the cross (Ferrão et al. 2013).
One of the main focuses of the Epamig/UFV/Embrapa breeding programs is to recommend a combination of Hybrid coffee seeds that avoid genetic incompatibility problems; are more drought-tolerant than clonal varieties; and are associated with rust resistance, maturation uniformity, and greater fruit size. Thus, by observing the results and disregarding H0915-1, which was allocated to the Robusta varietal group, the Hybrids H0911-2, H0919-2, H0922-1, H0912-1, H0921-1, and H0918-1 (Table 6) may be remarkable for the breeding program since they present the highest values of genetic dissimilarity.
Results revealed the great efficiency of technique used in the present study for the identification and genotyping of SNP markers, as well as the ability os these SNP to evaluate the genetic diversity. The most common molecular markers used in diversity studies in the genus Coffea are the Random Amplified Polymorphic DNA (Sera et al. 2003), Amplified Fragment Length Polymorphism (Prakash et al. 2005; Brito et al. 2010; Diola et al. 2011), and Simple Sequence Repeat (Prakash et al. 2005; Ferrão et al. 2013). A successful study has been carried out using SNP molecular markers, obtained by the genotyping-by-sequencing (GBS) approach (Ferrão et al. 2017). The authors investigated genomic selection models suitable for use in C. canephora. According to the authors, the results can be used as a basis for further studies on the genus Coffea and can be expanded for similar perennial crops (Ferrão et al. 2017). Another recent work, using SNP markers in coffee, obtained by the GBS technique, enabled a more comprehensive and significant study of the evolutionary history of the genus Coffea (Hamon et al. 2017).
Conclusions
The methodology of SNP identification and genotyping used in this study presents great potential to detect and select an expressive number of SNP markers.
The markers were efficient in evaluating the genetic diversity and population structure of C. canephora. Based on their analysis, promising crosses were selected within and between the varietal groups, according to the C. canephora breeding programs.
Due to the low genetic diversity of genotypes of the Conilon varietal group, when the objective is to increase the genetic base, genotypes with the highest values of genetic distance must be selected, and the variability of this group in the Germplasm Bank must be amplified by introducing new accessions to the Conilon population.
The highest values of genetic distances were observed between genotypes of Conilon and Robusta heterotic groups.
Hybrids with higher values of genetic distances can be selected, which are important for C. canephora breeding programs since they avoid genetic incompatibility problems.
References
Abatepaulo ARR, Caetano AR, Mendes CT Jr et al (2008) Detection of SNPs in bovine immune-response genes that may mediate resistance to the cattle tick Rhipicephalus (Boophilus) microplus. Anim Genet 39:328–329. https://doi.org/10.1111/j.1365-2052.2008.01713.x
Anderson CA, Pettersson FH, Clarke GM et al (2010) Data quality control in genetic case-control association studies. Nat Protoc 5:1564–1573. https://doi.org/10.1038/nprot.2010.116
Babova O, Occhipinti A, Maffei ME (2016) Chemical partitioning and antioxidant capacity of green coffee (Coffea arabica and Coffea canephora) of different geographical origin. Phytochemistry 123:33–39. https://doi.org/10.1016/j.phytochem.2016.01.016
Berthaud J (1986) Les resources génétique pour l’amélioration des caféiers africains diploides. Evaluation de la richesse génétique des populations sylvestres et de ses mécanismes organisateurs. Conséquences pour l’application. Université de Paris
Brito GG, Caixeta ET, Gallina AP et al (2010) Inheritance of coffee leaf rust resistance and identification of AFLP markers linked to the resistance gene. Euphytica 173:255–264. https://doi.org/10.1007/s10681-010-0119-x
Caetano AR (2009) Marcadores SNP: conceitos básicos, aplicações no manejo e no melhoramento animal e perspectivas para o futuro. Rev Bras Zootec 38:64–71. https://doi.org/10.1590/S1516-35982009001300008
Carvalho MCCG, Silva DCG (2010) Sequenciamento de DNA de nova geração e suas aplicações na genômica de plantas. Ciência Rural 40:735–744. https://doi.org/10.1590/S0103-84782010000300040
Cruz CD (2013) GENES—a software package for analysis in experimental statistics and quantitative genetics. Acta Sci Agron 35:271–276. https://doi.org/10.4025/actasciagron.v35i3.21251
Cubry P, Musoli P, Legnate H et al (2008) Diversity in coffee assessed with SSR markers: structure of the genus Coffea and perspectives for breeding. Genome 51:50–63. https://doi.org/10.1139/G07-096
Cubry P, De Bellis F, Pot D et al (2013) Global analysis of Coffea canephora Pierre ex Froehner (Rubiaceae) from the Guineo-Congolese region reveals impacts from climatic refuges and migration effects. Genet Resour Crop Evol 60:483–501. https://doi.org/10.1007/s10722-012-9851-5
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Davis AP, Govaerts R, Bridson DM, Stoffelen P (2006) An annotated taxonomic conspectus of the genus Coffea (Rubiaceae). Bot J Linn Soc 152:465–512. https://doi.org/10.1111/j.1095-8339.2006.00584.x
Denoeud F, Carretero-Paulet L, Dereeper A et al (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181–1184. https://doi.org/10.1126/science.1255274
Diniz LEC, Sakiyama NS, Lashermes P et al (2005) Analysis of AFLP markers associated to the Mex-1 resistance locus in Icatu progenies. Crop Breed Appl Biotechnol 5:387–393. https://doi.org/10.12702/1984-7033.v05n04a03
Diola V, de Brito GG, Caixeta ET et al (2011) High-density genetic mapping for coffee leaf rust resistance. Tree Genet Genomes 7:1199–1208. https://doi.org/10.1007/s11295-011-0406-2
Earl DA, VonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Ferrão LF, Caixeta ET, de Souza Fd et al (2013) Comparative study of different molecular markers for classifying and establishing genetic relationships in Coffea canephora. Plant Syst Evol 299:225–238. https://doi.org/10.1007/s00606-012-0717-2
Ferrão LFV, Caixeta ET, Pena G et al (2015) New EST–SSR markers of Coffea arabica: transferability and application to studies of molecular characterization and genetic mapping. Mol Breed 35:31. https://doi.org/10.1007/s11032-015-0247-z
Ferrão LFV, Ferrão RG, Ferrão MAG et al (2017) A mixed model to multiple harvest-location trials applied to genomic prediction in Coffea canephora. Tree Genet Genomes 13:95. https://doi.org/10.1007/s11295-017-1171-7
Gabriel SB, Schaffner SF, Nguyen H et al (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229. https://doi.org/10.1126/science.1069424
Garavito A, Montagnon C, Guyot R, Bertrand B (2016) Identification by the DArTseq method of the genetic origin of the Coffea canephora cultivated in Vietnam and Mexico. BMC Plant Biol 16:242. https://doi.org/10.1186/s12870-016-0933-y
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing
Gartner GAL, McCouch SR, Moncada MDP (2013) A genetic map of an interspecific diploid pseudo testcross population of coffee. Euphytica 192:305–323. https://doi.org/10.1007/s10681-013-0926-y
Gnirke A, Melnikov A, Maguire J et al (2009) Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27:182–189. https://doi.org/10.1038/nbt.1523
Gomez C, Dussert S, Hamon P et al (2009) Current genetic differentiation of Coffea canephora Pierre ex A. Froehn in the Guineo-Congolian African zone: cumulative impact of ancient climatic changes and recent human activities. BMC Evol Biol 9:1–19. https://doi.org/10.1186/1471-2148-9-167
Grandillo S (2014) Introgression libraries with wild relatives of crops. Genomics Plant Genet Resour 2:87–122. https://doi.org/10.1007/978-94-007-7575-6_4
Hamon P, Grover CE, Davis AP et al (2017) Genotyping-by-sequencing provides the first well-resolved phylogeny for coffee (Coffea) and insights into the evolution of caffeine content in its species. Mol Phylogenet Evol 109:351–361. https://doi.org/10.1016/j.ympev.2017.02.009
Kosman E, Leonard KJ (2005) Similarity coefficients for molecular markers in studies of genetic relationships between individuals for haploid, diploid, and polyploid species. Mol Ecol 14:415–424. https://doi.org/10.1111/j.1365-294X.2005.02416.x
Krzywinski M, Schein J, Birol I et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. https://doi.org/10.1101/gr.092759.109
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Lashermes P, Andrzejewski S, Bertrand B et al (2000) Molecular analysis of introgressive breeding in coffee (Coffea arabica L.). Theor Appl Genet 100:139–146
Lee W-P, Stromberg MP, Ward A et al (2014) MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS ONE 9:e90581. https://doi.org/10.1371/journal.pone.0090581
Leroy T, Marraccini P, Dufour M et al (2005) Construction and characterization of a Coffea canephora BAC library to study the organization of sucrose biosynthesis genes. Theor Appl Genet 111:1032–1041. https://doi.org/10.1007/s00122-005-0018-z
Liao P-Y, Lee KH (2010) From SNPs to functional polymorphism: the insight into biotechnology applications. Biochem Eng J 49:149–158. https://doi.org/10.1016/j.bej.2009.12.021
Marraccini P, Vinecky F, Alves GSC et al (2012) Differentially expressed genes and proteins upon drought acclimation in tolerant and sensitive genotypes of Coffea canephora. J Exp Bot 63:4191–4212. https://doi.org/10.1093/jxb/ers103
Metsalu T, Vilo J (2015) ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucl Acids Res 43:W566–W570. https://doi.org/10.1093/nar/gkv468
Musoli P, Cubry P, Aluka P et al (2009) Genetic differentiation of wild and cultivated populations: diversity of Coffea canephora Pierre in Uganda. Genome 52:634–646. https://doi.org/10.1139/G09-037
Neves LG, Davis JM, Barbazuk WB, Kirst M (2013) Whole-exome targeted sequencing of the uncharacterized pine genome. Plant J 75:146–156. https://doi.org/10.1111/tpj.12193
Neves LG, Davis JM, Barbazuk WB, Kirst M (2014) A high-density gene map of loblolly pine (Pinus taeda L.) based on exome sequence capture genotyping. G3 4:29–37. https://doi.org/10.1534/g3.113.008714
Ojopi EPB, Gregorio SP, Guimarães PEM et al (2004) O genoma humano e as perspectivas para o estudo da esquizofrenia. Rev Psiquiatr Clínica 31:9–18. https://doi.org/10.1590/S0101-60832004000100003
Pinto LA, Stein RT, Kabesch M (2008) Impact of genetics in childhood asthma. J Pediatr (Rio J) 84:S68–75. https://doi.org/10.2223/JPED.1781
Prakash NS, Combes M-C, Dussert S et al (2005) Analysis of genetic diversity in Indian robusta coffee genepool (Coffea canephora) in comparison with a representative core collection using SSRs and AFLPs. Genet Resour Crop Evol 52:333–343. https://doi.org/10.1007/s10722-003-2125-5
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Ren J, Sun D, Chen L et al (2013) Genetic diversity revealed by single nucleotide polymorphism markers in a worldwide germplasm collection of durum wheat. Int J Mol Sci 14:7061–7088. https://doi.org/10.3390/ijms14047061
Resende MDV, Lopes PS, Silva RL, Pires IE (2008) Seleção genômica ampla (GWS) e maximização da eficiência do melhoramento genético. Pesqui Florest Bras 56:63–77
Resende M, Caixeta E, Alkimim ER et al (2016) High-throughput targeted genotyping of Coffea Arabica and Coffea Canephora using next generation sequencing. California, San Diego, p 1
Sera T, Ruas PM, Ruas CDF et al (2003) Genetic polymorphism among 14 elite Coffea arabica L. cultivars using RAPD markers associated with restriction digestion. Genet Mol Biol 26:59–64. https://doi.org/10.1590/S1415-47572003000100010
Song J, Yang X, Resende MFR et al (2016) Natural allelic variations in highly polyploidy Saccharum complex. Front Plant Sci 7:1–18. https://doi.org/10.3389/fpls.2016.00804
Stacklies W, Redestig H, Scholz M et al (2007) pcaMethods a bioconductor package providing PCA methods for incomplete data. Bioinformatics 23:1164–1167. https://doi.org/10.1093/bioinformatics/btm069
Vieira LGE, Andrade AC, Colombo CA et al (2006) Brazilian coffee genome project: an EST-based genomic resource. Brazilian J Plant Physiol 18:95–108. https://doi.org/10.1590/S1677-04202006000100008
Yang W, Kang X, Yang Q et al (2013) Review on the development of genotyping methods for assessing farm animal diversity. J Anim Sci Biotechnol 4:2. https://doi.org/10.1186/2049-1891-4-2
Zhang P, Li J, Li X et al (2011) Population structure and genetic diversity in a rice core collection (Oryza sativa L.) investigated with SSR markers. PLoS ONE 6:e27565. https://doi.org/10.1371/journal.pone.0027565
Zhang J, Song Q, Cregan PB et al (2015) Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics 16:1–11. https://doi.org/10.1186/s12864-015-1441-4
Zhou L, Vega FE, Tan H et al (2016) Developing single nucleotide polymorphism (SNP) markers for the identification of Coffee Germplasm. Trop Plant Biol 9:82–95. https://doi.org/10.1007/s12042-016-9167-2
Acknowledgements
This work was financially supported by the Brazilian Coffee Research and Development Consortium (Consórcio Brasileiro de Pesquisa e Desenvolvimento do Café - CBP&D/Café), the Foundation for Research Support of the state of Minas Gerais (FAPEMIG), the National Council of Scientific and Technological Development (CNPq), and the National Institutes of Science and Technology of Coffee (INCT/Café).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Alkimim, E.R., Caixeta, E.T., Sousa, T.V. et al. High-throughput targeted genotyping using next-generation sequencing applied in Coffea canephora breeding. Euphytica 214, 50 (2018). https://doi.org/10.1007/s10681-018-2126-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-018-2126-2