
Abstract
Bermudagrass is a warm season grass widely cultivated for turf and fodder. Nonetheless, the grass has poor forage quality because animals that consume it fail to assimilate its organic matter efficiently. Thus, identification of the marker-trait association between simple sequence repeat (SSR) markers and forage-quality-related traits in diverse bermudagrass accessions would enable efficient selection of high forage quality bermudagrass cultivars. Association mapping of 8 forage-related-quality traits with 1474 markers was conducted in 60 diverse bermudagrass accessions from five geographical regions in China. Significant variations in eight phenotypic and physiological traits were observed among the 60 accessions. A total of 1474 alleles were amplified by 104 SSR primers. The average gene diversity and polymorphic information content for the study sample were 0.2097 and 0.1748 respectively. The clustering analysis suggested that geographic origin influenced genetic distances between accessions. A total of 76 markers significantly associated with traits at P < 0.01; 73 with a single trait and 3 with two traits each. Nevertheless, only 41 significant marker-trait associations (MTAs) were observed after Bonferroni test was separately conducted for each trait. Forty-one microsatellites had significant associations with 8 forage-quality-related traits. These markers provide a feasible means of genetically improving forage quality in bermudagrass after further authentication.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bermudagrass [Cynodon dactylon (L.), Pers] is a warm-season grass of the Poaceae family and Chloridinae subfamily of grasses (Casler and Duncan 2003). The Cynodon variety comprises of nine species and ten varieties which have been substantiated as important forage species; bermudagrass has been listed among the varieties which can be exclusively cultivated for forage (Wu and Taliaferro 2005).
In 1995, some scientists speculated that changes in the global environment would alter the quality and quantity of forage as well as grain mass production (Sinclair and Seligman 1995). Bermudagrass was initially mostly grown for pasture and hay in the tropical and subtropical areas. However, the recent global warming phenomenon has broadened the tropical belt, and some formerly temperate regions have become conducive for the growth of bermudagrass hence increasing the quantity of the grass grown in these areas. Obviously, there is a growing need to develop bermudagrass varieties with high forage quality required for animal husbandry and beef production. Bermudagrass is easily established via springs and shoots to produce astronomical biomass (Heldman 2003). Nonetheless, the grass has poor digestibility and hence low forage quality; animals feeding on bermudagrass are unable to efficiently digest and assimilate the organic matter of the grass due to the complex composition of its cell walls.
Forage quality refers to the animals’ effective and efficient conversion of feed into animal products (Fulgueira et al. 2007). Thus, animal performance, in production of produce such as milk, beef, and mutton, is a reflection of forage quality (Newman et al. 2009).
Digestibility and nitrogen content of grass are principle determinants of forage quality. The primary determinants of digestibility are the structural and cell-wall-associated components (CWCs) (Comont et al. Comont et al. 2013). Several studies of genetic diversity and genetic control of CWCs have been performed in principal crops such as ryegrass, rice, barley, and maize (Chen et al. 2012). The composition of cellular structures includes fats, proteins, fiber, minerals, and moisture. Thus, the main parameters to test during forage quality analysis are crude ash (CA), lignin, ADF, neutral detergent fiber (NDF), crude protein (CP), ammoniacal nitrogen, pH and dry matter (DM). The fiber content in forage comprises of ADF, NDF and acid detergent lignin (ADL). Both ADF and NDF contain cellulose and lignin, but only NDF has hemicellulose. ADF and NDF are critical indicators of forage quality because a negative correlation between the two and digestibility by livestock animals has been established (Xie et al. 2011). A direct proportion between lignin content and forage quality has also been documented. In fact, lipid deposition (lignin polymers) on the cell walls renders all warm-season grasses poor quality forage producers (Giordano et al. 2014). Unfortunately, less lignin in cells would weaken the plants and reduce the biomass while high lignin content would contribute to reduced digestibility. In maize (Zea mays), enzymes that control deposition of lignin in cell walls have been found to significantly influence digestibility and yield traits(Chen et al. 2010). Thus, one of the ways to enhance forage quality in bermudagrass would be the reduction of lignin content without compromising on its biomass (Wu and Taliaferro 2005). If researchers identify accessions with high forage-quality-related traits through marker-trait associations (MTAs), this improvement is possible.
Marker-trait association analysis has been done for agronomic traits in other plant species. They include tall fescue, rice, peanut, cotton and maize (Abdurakhmonov et al. 2008; Chen et al. 2010; Jiang et al. 2014; Lou et al. 2015; Zhang et al. 2014). A wide range of molecular markers has been utilized in association mapping studies. Although expressed sequence tags (EST-SSRs) are more transferable across species than genomic SSR markers and are critical for plant improvement (Mian et al. 2005), we used the latter because they were readily available. Genetic diversity of bermudagrass has been studied using SSR markers concerning cadmium tolerance (Xie et al. 2015) and seed yield (Wu et al. 2006). The objectives of our study were to: analyze the genetic diversity and genetic relationship of 60 bermudagrass accessions native to China; assess their phenotypic and physiological traits linked to forage quality and; identify marker-trait association in the 60 accessions.
Materials and methods
Plant materials
The 60 Bermuda grass accessions were collected from the turfgrass germplasm field at Wuhan Botanical Garden between September and October 2015. The different accessions had been initially collected from various geographic regions in China and planted in two different fields at Wuhan Botanical Garden turf grass using a randomly complete block design (Table S1; Fig. 1). Each of the accession was mowed with a manual rotary mower. All the mowed grass for each accession was weighed. The area that each grass type occupied was measured using a tape measure. The weight of total grass and the area would be utilized for calculation of biomass. About 20 grams of the mowed grass of each accession was collected in a paper envelope and transferred to the laboratory. The fresh weight (FW) of the each grass was recorded. The samples were then dried in the oven at 108 °C for 30 min and then at 70 °C until they achieved a constant weight which was registered as the dry weight (DW). The dry samples were pulverized with an electric pulveriser, between the months of December and January 2016, into a fine powder used for analysis of the forage-related contents.

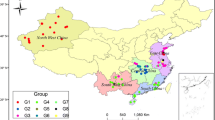
Distribution of the 7 cluster groups according to geographical regions. The map shows the five traditional regions of China. The map template was obtained from the dmaps.com website (http://www.d-maps.com/carte.php?num_car=17504andlang=en) and modified with Paint Windows 8.1 application. Arrows indicate the direction to and from main regions; on top of the map, arrows show direction along Northern China (North-west to North and North-east to North) while at the bottom part of the map, arrows show direction from Central China to Eastern China and Southern China (South-west China and South China). The five main geographical regions are North-west China, Central China, South-west China, South China and East China. The coloured triangles show each of the 7 groups identified by cluster analysis. Distribution of the coloured triangles shows the geographical origin of the members of each group
Phenotyping
The following phenotypic and physiological traits for every accession were measured: height (H), biomass (B), moisture content (MC), crude ash (CA), crude protein (CP), crude fat (CF), ADF and NDF.
For each genotype, six plants were randomly selected for height measurement with a ruler (cm). About 20 g of freshly mowed grass of each genotype was collected in a paper envelope, transported in the laboratory and dried in the oven. Moisture content was measured according to the following formula; Moisture content (MC) = [(FW − DW)/FW] × 100%.
Biomass (B) was calculated as total weight of grass divided by the area covered by the grass (tons/Ha). CP, CF, CA, ADF and NDF were analyzed according to the methods used in previous studies (Gul and Safdar 2009). All the phenotypes were analyzed for variation and significant correlation with each other using a two-tailed Pearson correlation test (n = 8, P < 0.05 and P < 0.01) conducted with IBM SPSS Statistics 24.0 (IBM. Corp 2014). A tow-tailed test was chosen because we could not predict the interaction between the traits and the data showed a normal distribution.
The results from the two fields had no significant difference. Nonetheless, we calculated the average for each phenotype for use in data analysis.
Genotyping
Total genomic DNA isolation for each accession was done with cetyl -trimethyl-ammonium-bromide method (CTAB) similar to that used in previous studies (Saghai Maroof et al. 1994; Xie et al. 2015). Resolution of the DNA’s concentration and quality was conducted with an ultraviolet (UV) spectrophotometer on 0.8% agarose gel (Xie et al. 2014). Polymerase chain reaction (PCR) was performed in a total volume of 10 µL containing 2 µL of 15 ng µL DNA template, 1 µL of 1 pmol µL−1 forward primer, 1 µL of pmol µL−1 reverse primer, 0.2 µL of 10 mMdNTPs (Pharmacia, America), 0.6 µM MgCl2, 1 U µL 1× Taq DNA polymerase and 3.95 µL H2O. The following procedure was used to carry out the PCR in a Mastercycler gradient PCR machine (Eastwin, China): 5 min at 94 °C, then 14 cycles of 20 s at 94 °C, 1 min at 58 °C, 30 s at 72 °C and a final extension step at 72 °C (Xie et al. 2015). The SSR markers (104 in total, from Department of Plant and Soil Sciences, Oklahoma State University obtained with Dr. Wu’s authorization) were screened in all the 60 Bermuda grass genotypes. These fluorescent dyes: HEX (green), TAM (yellow) and FAM (Blue), were used to label the 5′ end of the forward primers. Separation of the amplified fragments was achieved with an ABI 3730 DNA Sequencer (Applied Biosystem, Inc., Foster City, CA, USA). The GeneMarker 1.5 Software was used to label the alleles which were then checked twice manually to ensure accuracy. For each of the microsatellite, the presence or absence of a band was recorded as (1) and (0) respectively into a binary data matrix (Saha et al. 2005; Sun et al. 2015).
Population structure, kinship and genetic diversity
The Structure 2.3.4 software program was used to carry out a Bayesian model-based clustering analysis to infer a population structure to the 60 accessions (Pritchard et al. 2000). Ten independent simulations were conducted for K = 1 to K = 10 where K represents the number of assumed populations. For each simulation, 10,000 iterations were set as the burn-in time and 100,000 iterations of Markov Chain Monte Carlo (MCMC) were conducted with admixture and correlated frequency models, the best configuration for muted population structure (Falush et al. 2003). The method eliminated admixture of correlated allele frequencies and hence, the allocation of individual genotype to K subpopulations allowed for linkage equilibrium and validity of the Hardy–Weinberg equation within the population. The structure results were obtained via the structure harvester to determine the best K value, and a bar plot of the individuals with different colors indicating the posterior probability of their assignment to specific clusters was obtained (Earl and vonHoldt 2012). The most appropriate K value was determined by the posterior probability denoted as Ln P (D) and the delta K (Evanno et al. 2005; Pritchard et al. 2000; Porras-Hurtado et al. 2013). Moreover, the real value of K corresponds to the maximal value of L(K) generated by structure(Ciofi et al. 2002; Evanno et al. 2005; Vernesi et al. 2003; Zeisset and Beebee 2001).
A spatial pattern analysis of genetic diversity (SPAGeDi) program was used in the calculation of relative kinship among the 60 bermudagrass accessions. The software is tailor-made to use population genotypic data or mapped individuals’ spatial genetic structure (Hardy and Vekemans 2002). A pairwise kinship matrix was then made using the Loissele coefficient (Loiselle et al. 1995) and all the negative values between individuals were set to zero (Yu and Buckler 2006) because these individuals are more distant than random individuals (Yu et al. 2006).
The 60 bermudagrass accession were classified according to their geographical area of origin. These regions were East China, South China, South-west China, North-west China and Central China. The Powermarker 3.25 program was used to determine the extent of genetic variation in the five groups of accessions (Liu and Muse 2005). Both the polymorphic information content (PIC) and gene diversity were calculated (Nei 1972). The unweighted pair group method with arithmetic average (UPGMA) was used to construct a dendrogram based on Nei’s genetic distances. The genetic distances between groups, FST values, and linkage disequilibrium were also calculated with the structure analysis using Powermarker 3.25.
Association mapping
Among the 1474 alleles amplified by the 104 SSR primers, only 1326 with high PIC content were used for association analysis. Permutation test (1000 permutations, P < 0.01) to determine marker-trait association was done with the trait analysis by association, evolution and linkage (TASSEL 3.0), using the general linear model (GLM) (Bradbury et al. 2007). Association between markers and traits was determined by the P values while R2 referred to the percentage of total variation attributable to a marker. For each character, the critical P values for assessing the significance of SSR markers were calculated according to the false discovery rate (FDR), with FDR cut-off of 0.05 and the Bonferroni test using the Benjamini-Hochberg.xlsx (Benjamini and Hochberg 1995; Weinkauf 2012).
Results
Forage-quality-related trait variation
The summary statistics including the mean, range, standard deviation, variance and coefficient of variation (CV) were calculated in excel spread sheet for 8 traits in 60 accessions (Table 1). The CV was highest for crude protein (CP with 0.34) followed by Biomass (B with 0.33) crude fat (CF with 0.28) and moisture content (MC with 0.23). NDF and ADF had the least CV of 0.04 and 0.08 respectively. Height (H) and crude ash (CA) had relatively moderate CV of 0.20 and 0.19 respectively.
Pearson correlation coefficients of the phenotypic and physiological traits were calculated at the significance level of P < 0.01 and P < 0.05 (Table 2). The correlation coefficient of the traits ranged from r = −0.356 (between NDF and CP) to r = 0.475 (between CP and CF) with an average of r = 0.0595. Both positive and negative correlations were observed between the traits. Statistically significant positive correlations at P < 0.01 were found between B and H and CP and CF. Conversely, a statistically significant negative correlation at P < 0.01 was observed between CP and NDF. All statistically significant associations at P < 0.05 were negative; they occurred between B and MC, B and CP and NDF and CF (Table 2).
Population structure, kinship and genetic differentiation
Population structure
The population structure of the 60 bermudagrass accessions was derived using 1474 SSR markers and a model-based Structure 2.3.4 software. The possible number of groups (K) was set to range from 1 to 10, and 10 replications for each K value were performed to infer the population structure. The Ln P (D) value for each K value increased, nonetheless, the most significant increase occurred between K = 1 and K = 2 implying that the population comprised of two subpopulations and hence K was set to 2 (Fig. 2a). The highest L (K) value corresponded to the actual K value. The Q matrix obtained from running the K = 2 parameter was used to determine the membership probability for assigning accessions into their particular subpopulations when the probability was >0.75 (Fig. 2b). The majority of grasses had a membership probability higher than 0.9.

Population structure analysis of 60 bermudagrass accessions. GI and GII are the resultant structure groups. a. Magnitude of delta K for each K value (K represents the number of populations). b. Inferred population structure of the 60 bermudagrass accessions
Structure divided the 60 grass samples into two groups, GI and GII. GI had 56 individuals (93%) of all geographical regions except North-west China. All the 4 accessions from North-west China clustered in GII (Fig. 2b). The average distances or expected heterozygosity between individuals in GI was 0.1981 while that between individuals in GII was 0.1576.
Kinship
The population lacked kinship relationships; about 79% of the pairwise kinship coefficients were between 0 and 0.05 while about 13% were between 0.05 and 0.1 (Fig. 3). Thus, this approximation of kinship coefficient less than 0.05 represents the minimum relative relatedness among the samples; this portion is likely to affect association analysis.

The distribution of pair-wise relative kinship coefficients between the 60 bermudagrass accessions. Y axis shows percentage of distribution while X axis shows relative kinship
Cluster analysis
The 60 bermudagrass accessions fell into 7 distinct groups named G1–G7 (Fig. 4). Group 1 comprised of 7 individuals which further fell into two different subgroups. One of the subgroup contained 3 accessions from East China while the other contained 1 grass from East China, 1 from Central China and 2 from South-west China. Group 2 comprised of 22 accessions which fell into 2 distinct subgroups; one subgroup contained 4 kinds of grass from North-west China, 2 types of grass from East China and 1 grass-type from Central China. The other subgroup included 13 accessions from East China, 1 from South-west China and 1 from South China. Group 3 contained 2 accessions from Central China while Group 4 contained 4 accessions from South-west China. Group 5 comprised of 16 accessions from South China, Group 6 of 3 accessions from Central China and, Group 7 of 6 accessions, 5 from Central China and 1 from East China (Fig. 4).

Dendrogram of 60 Bermuda grass accessions achieved using 1474 SSR markers based on Nei’s genetic distance. Seven clusters: G1, G2, G3, G4, G5, G6, G7 bear different symbols as shown at the top right corner. Members collected from the same geographic regions clustered together. Individuals from adjacent regions were close to each other while those distant regions clustered in far-apart groups
Gene diversity of the entire collection of bermudagrass accessions
The total number of alleles obtained from the 1474 SSR loci scored for the 60 accessions was 2811 with an average of 1.91 alleles per marker. Major allele frequency (MAF) occurred at the range of 0.5–1.0 with an average of 0.856. The entire sample’s gene diversity was in the range of 0–0.5 with a mean gene diversity of 0.2097 (Table S2).
The range of polymorphic information content (PIC) for all the accessions was 0–0.375 while the average PIC was 0.1748. The heterozygosity of the entire sample was 0 while the number of observation of the markers ranged from 37 to 60. Of all the markers, 137 had a PIC value of zero. These and other 11 markers with little PIC were excluded in further analysis using Powermarker. 170 bands were high polymorphism alleles (PIC > 0.35) while 1167 alleles had moderate polymorphism (0.01 < PIC < 0.35) (Table S2).
Genetic relationships among 60 accessions grouped according to geographic origin
The statistics regarding gene diversity and PIC present at each locus in each population based on the geographic origin of bermudagrass accessions used in this study revealed an average gene diversity of 0.157 and an average PIC of 0.128. Both Central China and East China had the highest gene diversity and PIC of 0.184 and 0.150 respectively. South China closely followed with gene diversity of 0.162 and PIC of 0.133. North-west China population had the least gene diversity and PIC. The average gene diversity and PIC for each one of the population were as follows: Central China = 0.184 and 0.150, North-west China = 0.103 and 0.082, East China = 0.184 and 0.150, South-west China = 0.152 and 0.123 and South China = 0.162 and 0.133 (Table 3).
Genetic distances, FST values and linkage disequilibrium (LD)
North-west china and South-west china were the furthest distance apart with a pairwise genetic distance of 0.1536 followed by North-west china and South china with 0.1424. The least genetic distance was observed between East China and Central China (0.0276) followed by Central China and South-west China (0.0313) (Table 4). Nei’s distance between the two structure populations (GI and GII) was 0.1763.
FST values were calculated via structure analysis to show genetic differentiation of the subpopulations (Table 5). The maximum FST value was 0.422 for South-west China followed by 0.406 for Central China. The minimum FST value was 0.012 for South China. East China and North-west China had FST values of 0.117 and 0.043 respectively.
A total of 1335 pairs of markers were found to be in disequilibrium after pairwise linkage disequilibrium at the allele level. Of these pairs, 746 were in complete LD (D’ = 1) while according to r2, 25 pairs were in perfect LD (r2 = 1).The percentage of significant SSR loci pairs in LD was 22% and 33% at r2 > 0.1 was and at r2 > 0.05 respectively (Table S3).
Marker-trait association
A total of 8400 MTAs between 1337 markers and the 8 traits were observed. However, only 76 of these marker-trait association were significant at P < 0.01. Most markers had a significant association with both H and CA, 13 markers each, followed by MC with 11 markers and ADF with 10 markers. B had 5 MTAs, the least among the 8 traits. Three of the markers had significant MTAs at P < 0.01 with more than one trait; these were M109 with B and H, M1415 with H and B and M890 with CF and CP (Table S4). Nonetheless, no marker had a significant association with more than two traits at P < 0.01.
Only 41 markers had significant marker-trait association after independent Bonferroni test for each character. The following marker-trait associations were observed: 4 markers associated with ADF, 5 with B, 10 with CA, 5 with CF, 3 with CP, 7 with H, 4 with MC and 3 with NDF (Table 6).
The percentage of variation attributable to a marker (R2) ranged between 9 and 23%. The highest was observed in the marker-trait association between M1170 and CA and the least between the associations of both M1418 and M83 with B. The R2 for multiple MTAs were moderate ranging between 11 and 14%. The highest R2 values for each of the other traits were: CF 16%, CP 17%, ADF and NDF 17%, MC 18% and H 18% (Table S4).
Discussion
The large variation in forage-quality-related traits observed among accessions as well as the significant correlations between these traits reflects the rich phenotypic and physiological trait diversity among the 60 bermudagrass accessions. These findings imply that our study sample qualified for marker-trait association mapping for different forage-quality-related traits.
Plant scientists have identified CV as a critical indicator to guide the selection procedure for target phenotypic variants in breeding programs (Lou et al. 2015) since it is an indicator of variability of quantitative traits. CP, B and CF with 0.34, 0.33, and 0.28 CV respectively could be useful in selection breeding programs purposing to increase the B and CP and CF in forage. Although MC, H, CA, NDF and ADF had lower CV in that order, they reveal that the study sample has diversity which can gradually sum up to produce heterosis.
The results of the phenotypic analysis indicate that reducing B could increase both MC and CP and consequently improve forage quality since B negatively correlated to MC and CP. They also imply that reducing NDF would result in increased CF and CP. However, our study seeks to improve forage quality without interfering with B and the latter observation is more valuable and suitable for application in breeding programs. Since NDF contains lignin, these results support the hypothesis that reduction of lignin content would increase forage quality by increasing crude fat and crude protein. Our results are consistent with similar results which showed NDF to have a negative correlation with CP, CP to have a positive relationship with CF and, although not significant, CA to have a positive relation with CP (Gharehshekhlou et al. 2012; Sluiter et al. 2008; Xie et al. 2009).
The accessions used in this study grew under the same environmental conditions and were harvested at the same time. Therefore, the variation in the phenotypic and physiological traits cannot be attributable to environmental factors, soil characteristics or time of harvest. The difference we observed could only result from genetic differences among the accessions. We found significant genetic variations between accessions which are consistent with a similar study on bermudagrass accessions collected from different regions in China (Xie et al. 2014, 2015). Our study sample, therefore, represents a germplasm with the potential for use in future breeding programs. It suggests the possibility of controlling the forage quality via regulation of CP, B and CF contents which have high CV.
Furthermore, population structure, kinship and genetic diversity analysis results confirms the reliability of our study for use in breeding strategies aimed at improving forage quality in bermudagrass. The structure results gave two groups and hence, one covariate which has been suggested to be inefficient in controlling stratification (Mezmouk et al. 2011). Thus, it was prudent to use a reliability test which depended on the distance between the structure-generated outputs by evaluating the similarity of the Q matrices. However, in a previous study, the neighbour-joining tree using a subset of 342 lines only showed a consistency between structure-derived groups and clustering when K = 2 and K = 3 but showed a vague trend for more K values (Camus-Kulandaivelu et al. 2006). These findings confirm previous observations that combining structure’s results and distances between clusters generated on the neighbour-joining tree improves the reliability of the association analysis study.
In studies of genetic diversity, researchers have come to a concession that different types of markers yield different results of population structure when analysed with the structure software. Nonetheless, microsatellite markers have been identified to perform better than amplified fragment length polymorphisms (AFLPs) markers (Evanno et al. 2005). Furthermore, proper sampling of individuals and markers alike has been determined to influence the outcome of the groups produced by the structure software (Evanno et al. 2005). Thus, the use of SSR markers plus the selection of diverse accessions makes our association mapping study reliable.
A remarkable consistency, with negligible discrepancies, between the distribution of accessions according to geographic location, structure results and cluster analysis was observed. The population structure derived from structure analysis showed an association between population structure and geographical origin which was consistent with similar studies on bermudagrass collected from China (Xie et al. 2014, 2015).
All the accessions that fell under structure’s GII clustered together in Nei’s dendrogram and shared a common geographical origin, that of North-west China. Furthermore, some of the cluster groups (G3, G4, G5 and G6) exclusively contained members from the same geographical origin. In addition, accessions with furthest distance apart clustered away from each other as shown for North-west China and South-west China. The clustering together of accessions collected from similar or adjacent regions could imply a strong correlation between genetic diversity and geographic location (Li et al. 2011). The results of our study closely resonate with previous studies on genetic diversity of bermudagrass collected in China using both ISSR and SSR markers (Wang et al. 2013), and SSR markers only (Xie et al. 2014, 2015).
The pairwise kinship matrix confirmed that the accessions were relatively distant. This phenomenon could be attributable to the diversity of the genotypes and, the fact that accessions were collected from different regions eliminating the possibility of analyzing similar genotypes.
The genetic diversity of the entire sample collection was 0.2097 and lower than the 0.2270 previously reported for 120 bermudagrass accessions. The PIC was 0.1748 and also lower than the 0.1894 reported for 120 bermudagrass accessions (Xie et al. 2015). Our study sample has less genetic diversity than the one used in the previous study utilizing a similar number of markers. Thus, these discrepancies could be due to a large number of markers used on a small population (60 samples).
Other than the PIC values, FST values can be used to define the genetic diversity of a population; they can determine the standardized population distance between populations (Dang et al. 2016), due to their relationship with allele frequencies and genetic diversity (Jakobsson et al. 2013). According to the FST values, Central China and South-west China are highly differentiated from the other groups. East China, North-west China and South China are less differentiated. These results imply that Central China and South-west China contributed the least genetic material in GI while East China and South China contributed the most genetic material. Although North-west China (FST = 0.043) is likely to exchange genetic material, it is physically isolated from the other groups hindering exchange of genetic material. Besides it has the furthest genetic distance from either of the groups and since structure’s GI1 exclusively contains accessions from this geographical origin, it has low genetic diversity.
The genetic distances show East China and Central China to be significantly closer (0.0276). Genetic diversity was also highest in East China and Central China but lowest in North-west China. North-west China is, however, genetically closer to East China (0.1250) than to Central China (0.1356) (Table 4). These results coincide with the clustering results shown in Nei’s dendrogram where East China and Central China appear close to each other at the extreme ends of the dendrogram with North-west China being closer to East China than to Central China. Since the two groups with the highest genetic diversity and the shortest genetic distance apart fell under structure’s GI, it is more diverse than GII because the distinct groups can exchange genetic material. Besides, the average genetic distance between GI members was higher than that between GII members implying that GII members were readily exchanging genetic materials than GI members. These results could be attributable to exchange of genetic material between adjacent members since bermudagrass is an outcrossing variety. Members of GII were physically separated from the other groups promoting inbreeding and hence the low genetic diversity. GI, therefore, contained rich genetic diversity which could be utilized in breeding programs to improve forage quality of bermudagrass.
Although the percentage of markers in LD was high in terms of D’(55% in complete LD), we cannot rely on this criterion because it is inflated when one allele is rare or the sample size is small. However, perfect LD (defined by r2) is reliable for association mapping because it will persist until recombination occurs and loci in LD can remain in strong LD for a very long time (Montgomery Slatkin 2008). Thus, recombination events that occurred in the past can be exploited through association mapping. The percentage of significant SSR loci pairs in LD at r2 > 0.1 (22%) and r2 > 0.05 (33%) observed in our study is similar to that reported for cotton (22%), and relatively higher than reported for sorghum (8.7%), and maize (10%) (Abdurakhmonov et al. 2009; Hamblin et al. 2004; Remington et al. 2001). Higher percentages of SSRs in LD have been reported for durum wheat (49–56%) and cultivated barley germplasm (45–100%) (Kraakman et al. 2004; Maccaferri et al. 2005). Bermudagrass being an outcrossing breed, we expected it to have lower LD than inbreeding varieties such as sorghum since LD in outbreeding varieties is usually low. Compared to maize, which is a highly outbreeding crop, we can consider the bermudagrass in our study to have high LD. On the contrary, the bemudagrass has a moderate LD as compared to wheat and barley. Nonetheless, it qualifies for association mapping although with low resolution. The presence of the moderate LD could imply the presence of rare alleles which could be caused by mutation.
Through association mapping the rare alleles linked to markers associated with forage-quality-related traits could be identified and exploited in breeding to improve forage quality. The ease of accessibility to many molecular markers and developed mapping populations recently has enabled association mapping to identify the qualitative trait loci (QTLs) for agronomic traits (Ravi et al. 2011; Selvaraj et al. 2009). Several scientists have emphasized the need to control association mapping studies to avoid spurious marker-trait association (Flint-Garcia et al. 2005; Yu et al. 2011). Both genetic linkage and population structure have the capacity to cause marker-trait association. Furthermore, individuals from different geographical locations subjected to association analysis may possess a population structure or kinship ties as a consequence of selection or adaptation to a local habitat (Yu and Buckler 2006). Thus, it is crucial to analyze the population structure of a sample intended for association mapping studies since with time, inbreeding and evolution have inevitably created population structures and kinship ties (Yu and Buckler 2006). It is evident from the structure results that the 60 bermudagrass accessions used for this study had a population structure but lacked kinship relatedness.
In association analysis, two traditional models are used; these are the general linear model (GLM) and the mixed linear model (MLM). Both account for population structure, however, the latter also accounts for kinship while the former does not. Although using both models limits the number of false positives (Yu et al. 2006), we used the GLM model because our population lacked kinship ties but had a distinct population structure. Besides, most of the MTAs identified with the MLM model become statistically insignificant after marker-trait association analysis with the GLM model (Chen et al. 2010).
A total of 41 significant MTAs were observed. The trait with the highest number of MTAs was CA with 10 while the least was CP and NDF each with 3 MTAs. Of the 10 MTAs involving CA, two pairs appeared close to each other and could belong to the same locus; these were M915 and M916 as well as M1170 and M1171. However, CA had no significant correlation with the other traits and this information could be irrelevant for application in selection procedures. Height had 7 MTAs of which M920 and M921 are close implying location in similar loci. No other close markers involved in MTAs of the same trait were observed. Thus, it is evident that different markers significantly associated with a particular trait as no marker had significant association with more than one trait.
In our study, three markers significantly associated with correlated traits (prior to Bonferroni correction) indicating presence of a pleiotropic gene or tight linkage of several genes. These markers were M890 associated to CP and CF (which had a positive correlation) and M1415 and M190 both associating with B and H (which also had a positive correlation). B was also correlated to MC and CP while CP correlated to NDF. Thus, the three loci positions contained a gene that could control all these phenotypes or genes which controlled each of these phenotypes were tightly linked. Fine-mapping of the chromosomal regions in these loci is imperative for the discernment of the actual genetic control of these congruent traits (Li et al. 2012). If experts could design markers for these congruent traits in the particular locus, the implementation of potent marker-assisted selection (MAS) for enhancing forage quality in bermudagrass would be possible.
Unfortunately, the low resolution of MTAs, low proportion of phenotypic variation explained by the QTLs (<10), and limited success in validating QTLs in different scenarios limits the implementation of MAS (Neale et al. 2002). Besides, MTAs with R2 greater than 10% show high heritability although they might be difficult to identify for congruent traits (Wang et al. 2012). This limitation arises because QTLs’ effect is highly dependent on the breeding strategy of a plant (inbreeding or outbreeding) as well as the sum of genes with little control over the expression of a qualitative trait (Guimarães et al. 2007). Bermudagrass is a highly outcrossing species (Wu and Taliaferro 2005) and it is, therefore, impossible to use recombination-based methods for position cloning of genes (Salvi et al. 2002). Notwithstanding, fine-mapping would be required for practical use of the identified significant MTAs.
In our study, the average variation attributable to the marker ranged from 11% for B to 19% for CA implying feasibility of MAS. Moreover, since we identified population subdivision in our study, we can rule out the possibility of LD being a result of unrecognized population subdivision; the markers in significant LD were indeed linked suggesting that linkage was the primary cause of LD. Nevertheless, the number of statistically significant MTAs is quite low and unsuitable (76 at P < 0.01 and 41 after Benjamini–hochberg correction) for direct use in MAS. Successful MAS based on our results would require the selective choice of parents for QTL mapping and elaborate crosses to verify the effectiveness of marker alleles critical for developing high forage quality bermudagrass cultivars.
Conclusion
In summary, our study showed diversity in phenotypic variation in ADF, B, CA, CF, CP, H, MC and NDF among the 60 bermudagrass accessions intended for association mapping for forage quality. The clustering analysis revealed that genetic distance between accessions was affected by geographical origin. The individuals fell into two subpopulations, and the entire sample lacked kinship relatedness. Seventy-six MTAs were significant at P < 0.01 while only forty-one were significant after Bonferroni correction of the P values. Three of the markers associated with two correlated forage-related-traits and could play a meaningful role in the determination of forage quality. The results of our study can be used for fine mapping to establish the exact contribution of the genes in loci positions of these markers for use in MAS to enhance forage quality of bermudagrass.
Abbreviations
- ADF:
-
Acid detergent fiber
- ADL:
-
Acid detergent lignin
- AFLPs:
-
Amplified fragment length polymorphisms
- B:
-
Biomass
- CA:
-
Crude ash
- CF:
-
Crude fat
- CP:
-
Crude protein
- CTAB:
-
Cetyl-trimethyl-ammonium-bromide
- CV:
-
Coefficient of variation
- CWCs:
-
Cell-wall-associated components
- DM:
-
Dry matter
- EST:
-
Expressed sequence tags
- FDR:
-
False discovery rate
- GLM:
-
General liner model
- H:
-
Height
- LD:
-
Linkage disequilibrium
- MAF:
-
Major allele frequency
- MC:
-
Moisture content
- MTAs:
-
Marker-trait associations
- MLM:
-
Mixed linear model
- NDF:
-
Neutral detergent fiber
- PIC:
-
Polymorphic information content
- QTLs:
-
Qualitative trait loci
- SSR:
-
Simple sequence repeats
- TDN:
-
Total digestible nutrients
References
Abdurakhmonov IY, Kohel RJ, Yu JZ, Pepper AE, Abdullaev AA, Kushanov FN, Salakhutdinov IB, Buriev ZT, Saha S, Scheffler BE, Jenkins JN, Abdukarimov A (2008) Molecular diversity and association mapping of fiber quality traits in exotic G. hirsutum L. germplasm. Genomics 92:478–487. doi:10.1016/j.ygeno.2008.07.013
Abdurakhmonov IY, Saha S, Jenkins JN, Buriev ZT, Shermatov SE, Scheffler BE, Pepper AE, Yu JZ, Kohel RJ, Abdukarimov A (2009) Linkage disequilibrium based association mapping of fiber quality traits in G. hirsutum L. variety germplasm. Genetica 136:401–417. doi:10.1007/s10709-008-9337-8
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. doi:10.2307/2346101
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. doi:10.1093/bioinformatics/btm308
Camus-Kulandaivelu L, Veyrieras JB, Madur D, Combes V, Fourmann M, Barraud S, Dubreuil P, Gouesnard B, Manicacca D, Charcosset A (2006) Maize adaptation to temperate climate: relationship between population structure and polymorphism in the Dwarf8 Gene. Genetics 172:2449–2463. doi:10.1534/genetics.105.048603
Casler MD, Duncan RR (2003) Turfgrass biology, genetics, and breeding. Wiley, New Jersey
Chen Y, Zein I, Brenner EA, Andersen JR, Landbeck M, Ouzunova M, Lübberstedt T (2010) Polymorphisms in monolignol biosynthetic genes are associated with biomass yield and agronomic traits in European maize (Zea mays L.). BMC Plant Biol 10:12. doi:10.1186/1471-2229-10-12
Chen X, Min D, Yasir TA, Hu Y-G (2012) Genetic diversity, population structure and linkage disequilibrium in elite Chinese winter wheat investigated with SSR markers. PLoS ONE 7:e44510. doi:10.1371/journal.pone.0044510
Ciofi C, Milinkovitch MC, Gibbs JP, Caccone A, Powell JR (2002) Microsatellite analysis of genetic divergence among populations of giant Galápagos tortoises. Mol Ecol 11:2265–2283. doi:10.1046/J.1365-294X.2002.01617.X
Comont D, Winters A, Gomez LD, McQueen-Mason SJ, Gwynn-Jones D (2013) Latitudinal variation in ambient UV-B radiation is an important determinant of Lolium perenne forage production, quality, and digestibility. J Exp Bot 64:2193–2204. doi:10.1093/jxb/ert077
Dang X, Liu E, Liang Y, Liu Q, Breria CM (2016) QTL detection and elite alleles mining for stigma traits in Oryza sativa by association mapping. Front Plant Sci 7:1–14. doi:10.3389/fpls.2016.01188
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. doi:10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Falush D, Stephens M, Pritchard J (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. doi:10.1111/j.1471-8286.2007.01758.x
Flint-Garcia SA, Thuillet AC, Yu J, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44:1054–1064. doi:10.1111/j.1365-313X.2005.02591.x
Fulgueira CL, Amigot SL, Gaggiotti M, Romero LA, Basílico JC (2007) Forage quality: techniques for testing. Fresh Prod 1:121–131
Gharehshekhlou HR, Rasouli B, Ghotbi AA, Amiri B (2012) Forage quality analysis of Dactylic glomerata, Onobrychis sativa and Setaria galauca in North of Iran by different methods. Res J Appl Sci Eng Technol 4:1962–1966
Giordano A, Liu Z, Panter SN, Dimech AM, Shang Y, Wijesinghe H, Fulgueras K, Ran Y, Mouradov A, Spangenberg GC (2014) Reduced lignin content and altered lignin composition in the warm season forage grass Paspalum dilatatum by down-regulation of a Cinnamoyl CoA Reductase Gene. Transgenic Res 23:503–517. doi:10.1007/s11248-014-9784-1
Guimarães EP, Ruane J, Scherf BD, Sonnino A, Dargie JD (2007) Marker-assisted selection: current status and future perspectives in crops, livestock, forestry and fish. Food Agric Org 127:1–494
Gul S, Safdar M (2009) Proximate composition and mineral analysis of cinnamon. Pak J Nutr 8:1456–1460. doi:10.3923/pjn.2009.1456.1460
Hamblin MT, Mitchell SE, White GM, Gallego J, Kukatla R, Wing RA, Paterson AH, Kresovich S (2004) Comparative population genetics of the panicoid grasses: sequence polymorphism, linkage disequilibrium and selection in a diverse sample of Sorghum bicolor. Genetics 167:471–483
Hardy OJ, Vekemans X (2002) SPAGeDI: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620. doi:10.1046/j.1471-8286.2002.00305.x
Heldman D (2003) Encyclopedia of agricultural, food, and biological engineering. Marcel Dekker, New York
IBM. Corp. (2014) IBM SPSS Statistics for Windows, Version 24.0. Armonk, NY
Jakobsson M, Edge MD, Rosenberg NA (2013) The relationship between FST and the frequency of the most frequent allele. Genetics 193:515–528. doi:10.1534/genetics.112.144758
Jiang H, Huang L, Ren X, Chen Y, Zhou X, Xia Y, Huang J, Lei Y, Yan L, Wan L, Liao B (2014) Diversity characterization and association analysis of agronomic traits in a Chinese peanut (Arachis hypogaea L.) mini-core collection. J Integr Plant Biol 56:159–169. doi:10.1111/jipb.12132
Kraakman ATW, Niks RE, Van den Berg PMMM, Stam P, Van Eeuwijk FA (2004) Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 446:435–446. doi:10.1534/genetics.104.026831
Li H, Liu L, Lou Y, Hu T, Fu J (2011) Genetic diversity of Chinese natural bermudagrass (Cynodon dactylon) germplasm using ISSR markers. Sci Hortic 127:555–561. doi:10.1016/j.scienta.2010.12.001
Li X, Yan W, Agrama H, Jia L, Jackson A, Moldenhauer K, Yeater K, McClung A, Wu D (2012) Unraveling the complex trait of harvest index with association mapping in rice (Oryza sativa L.). PLoS ONE 7:e29350. doi:10.1371/journal.pone.0029350
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129. doi:10.1093/bioinformatics/bti282
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Lou Y, Hu L, Chen L, Sun X, Yang Y, Liu H, Xu Q (2015) Association analysis of simple sequence repeat (SSR) markers with agronomic traits in tall fescue (Festuca arundinacea Schreb.). PLoS ONE 10:e0133054. doi:10.1371/journal.pone.0133054
Maccaferri M, Sanguineti MC, Noli E, Tuberosa R (2005) Population structure and long-range linkage disequilibrium in a durum wheat elite collection. Mol Breed 15:271–289. doi:10.1007/s11032-004-7012-z
Mezmouk S, Dubreuil P, Bosio M, Décousset L, Charcosset A, Praud S, Mangin B (2011) Effect of population structure corrections on the results of association mapping tests in complex maize diversity panels. Theor Appl Genet 122:1149–1160. doi:10.1007/s00122-010-1519-y
Mian MA, Saha MC, Hopkins AA, Wang ZY (2005) Use of tall fescue EST-SSR markers in phylogenetic analysis of cool-season forage grasses. Genome 48:637–647. doi:10.1139/g05-029
Montgomery S (2008) Linkage disequilibrium-understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9:477–485. doi:10.1038/nrg2361
Neale DB, Sewell MM, Brown GR (2002) Molecular dissection of the quantitative inheritance of wood property traits in loblolly pine. Ann For Sci 59:595–605. doi:10.1051/forest:2002045
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292. doi:10.1086/282771
Newman YC, Adesogan AT, Vendramini J, Sollenberger L (2009) Defining forage quality 1–5. The Texas A & M university system. http://publications.tamu.edu/FORAGE/PUB_forage_Defining%20Forage%20Quality.pdf. Accessed 20 Sept 2016
Porras-Hurtado L, Ruiz Y, Santos C, Phillips C, Carracedo A, Lareu MV (2013) An overview of STRUCTURE: applications, parameter settings, and supporting software. Front Genet 4:1–13. doi:10.3389/fgene.2013.00098
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. doi:10.1016/0379-0738(94)90222-4
Ravi K, Vadez V, Isobe S, Mir RR, Guo Y, Nigam SN, Gowda MV, Radhakrishnan T, Bertioli DJ, Knapp SJ, Varshney RK (2011) Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut (Arachis hypogaea L.). Theor Appl Genet 122:1119–1132. doi:10.1007/s00122-010-1517-0
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci U S A 98:11479–11484. doi:10.1073/pnas.201394398
Saghai Maroof MA, Biyashev RM, Yang GP, Zhangf Q, Allard RW (1994) Extraordinarily polymorphic microsatellite DNA in barley: species diversity, chromosomal locations, and population dynamics. Popul Biol 91:5466–5470. doi:10.1073/pnas.91.12.5466
Saha MC, Mian R, Zwonitzer JC, Chekhovskiy K, Hopkins AA (2005) An SSR- and AFLP-based genetic linkage map of tall fescue (Festuca arundinacea Schreb.). Theor Appl Gene 110:323–336. doi:10.1007/s00122-004-1843-1
Salvi S, Tuberosa R, Chiapparino E, Maccaferri M, Veillet S, van Beuningen L, Isaac P, Edwards K, Phillips RL (2002) Toward positional cloning of Vgt1, a QTL controlling the transition from the vegetative to the reproductive phase in maize. Plant Mol Biol 48:601–613. doi:10.1023/A:1014838024509
Selvaraj MG, Schubert AM, Ayers JL, Baring MR (2009) Identification of QTLs for pod and kernel traits in cultivated peanut by bulked segregant analysis. Electron J Biotechnol. doi:10.2225/vol12-issue2-fulltext-13
Sinclair TR, Seligman NG (1995) Global environment change and simulated forage quality of wheat I. Nonstressed conditions. Field Crop Res 40:19–27. doi:10.1016/0378-4290(94)00091-P
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D (2008) Determination of ash in biomass laboratory analytical procedure (LAP). Nrel/Tp-510-4262226: 18
Sun X, Du Z, Ren J, Amombo E, Hu T, Fu J (2015) Association of SSR markers with functional traits from heat stress in diverse tall fescue accessions. BMC Plant Biol 15:1–13. doi:10.1186/s12870-015-0494-5
Vernesi C, Crestanello B, Pecchioli E, Tartari D, Caramelli D, Hauffe H, Bertorelle G (2003) The genetic impact of demographic decline and reintroduction in the wild boar (Sus scrofa): a microsatellite analysis. Mol Ecol 12:585–595. doi:10.1046/J.1365-294X.2003.01763.X
Wang M, Jiang N, Jia T, Leach L, Cockram J, Waugh R, Ramsay L, Thomas B, Luo Z (2012) Genome-wide association mapping of agronomic and morphologic traits in highly structured populations of barley cultivars. Theor Appl Genet 124:233–246. doi:10.1007/s00122-011-1697-2
Wang Z, Liao L, Yuan X, Guo H, Guo A, Liu J (2013) Genetic diversity analysis of Cynodon dactylon (bermudagrass) accessions and cultivars from different countries based on ISSR and SSR markers. Biochem Syst Ecol 46:108–115. doi:10.1016/j.bse.2012.09.001
Weinkauf M (2012) BenjaminiHochberg.xlsx. Version 1.1
Wu Y, Taliaferro CM (2005) Bermudagrass. In: Singh RJ, Jauhar PP (eds) Genetic resources, chromosome engineering, and crop improvement. Taylor & Francis, Florida, pp 229–275
Wu YQ, Taliaferro CM, Martin DL, Goad CL, Anderson JA (2006) Genetic variability and relationships for seed yield and its components in Chinese Cynodon accessions. Field Crop Res 98:245–252. doi:10.1016/j.fcr.2006.02.003
Xie HL, Ji HQ, Liu ZH, Tian GW, Wang CL, Hu YM, Tang JH (2009) Genetic basis of nutritional content of stover in maize under low nitrogen conditions. Euphytica 165:485–493. doi:10.1007/s10681-008-9764-8
Xie J, Kong X, Chen J, Hu B, Wen P, Zhuang J, Bao J (2011) Mapping of quantitative trait loci for fiber and lignin contents from an interspecific cross Oryza sativa × Oryza rufipogon. J Zhejiang Univ Sci B 12:518–526. doi:10.1631/jzus.B1000299
Xie Y, Luo H, Hu L, Sun X, Lou Y, Fu J (2014) Classification of genetic variation for cadmium tolerance in Bermudagrass [Cynodon dactylon (L.) Pers.] using physiological traits and molecular markers. Ecotoxicology 23:1030–1043. doi:10.1007/s10646-014-1247-1
Xie Y, Sun X, Ren J, Fan J (2015) Genetic diversity and association mapping of cadmium tolerance in bermudagrass [Cynodon dactylon (L.) Pers.]. Plant Soil 390:307–321. doi:10.1007/s11104-015-2391-y
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160. doi:10.1016/j.copbio.2006.02.003
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208. doi:10.1038/ng1702
Yu X, Bai G, Luo N, Chen Z, Liu S, Liu J, Warnke SE, Jiang Y (2011) Association of simple sequence repeat (SSR) markers with submergence tolerance in diverse populations of perennial ryegrass. Plant Sci 180:391–398. doi:10.1016/j.plantsci.2010.10.013
Zeisset I, Beebee TJ (2001) Determination of biogeographical range: an application of molecular phylogeography to the European pool frog Rana lessonae. Proc R Soc London B Biol Sci 268:933–938. doi:10.1098/rspb.2001.1600
Zhang P, Liu X, Tong H, Lu Y, Li J (2014) Association mapping for important agronomic traits in core collection of rice (Oryza sativa L.) with SSR markers. PLoS ONE 9:e111508. doi:10.1371/journal.pone.0111508
Acknowledgement
This research was financially supported by the Young Scientists Fund of the National Natural Science Foundation of China (Grant Number 31502009) and National Natural Science Foundation of China (Grant Number 31672482).
Author information
Authors and Affiliations
Contributions
XY and JF conceived and designed the experiment. MMG performed the research, analyzed data and wrote the manuscript. JF and XY revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gitau, M.M., Fan, J., Xie, Y. et al. Genetic diversity and association mapping of forage quality in diverse bermudagrass accessions. Euphytica 213, 234 (2017). https://doi.org/10.1007/s10681-017-2024-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-2024-z