
Abstract
Soybean research has found that nodule traits, especially nodule biomass, are associated with N2 fixation ability. Two genotypes, differing in nodule number per plant and individual nodule weight, KS4895 and Jackson, were mated to create 17 F3- and 80 F5-derived RILs. The population was mapped with 664 informative markers with an average distance of less than 20 cM between adjacent markers. Nodule traits were evaluated in 3-year field trials. Broad-sense heritability for nodule number (no. plant−1), individual nodule dry weight (mg nodule−1), individual nodule size (mm nodule−1), and total nodule dry weight (g plant−1) was 0.41, 0.42, 0.45, and 0.27, respectively. Nodule number was negatively correlated with individual nodule weight and size. Nodule number, individual nodule weight, and size are major components which likely contributed to increased total nodule weight per plant. Composite interval mapping (CIM) identified eight QTLs for nodule number with R2 values ranging from 0.14 to 0.20. Multiple interval mapping (MIM) identified two QTLs for nodule number, one of which was located close to the QTL identified with CIM. Six QTLs for individual nodule weight were detected with CIM, and one QTL was identified with MIM. For nodule size, CIM identified seven QTLs with R2 values ranging from 0.14 to 0.27. Five QTLs for total nodule weight were detected with CIM, one of which was located close to a QTL identified with MIM. These results document the first QTL information on nodule traits in soybean from field experiments utilizing a dense, complete linkage map.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A fundamental advantage of legume crops in agricultural systems is the legume’s ability to supply much of its own nitrogen needs through symbiotic nitrogen fixation. The advantage of legume crops stems from the large amounts of nitrogen required by a productive crop and from the expense of nitrogen fertilizer (Purcell 2009). Soybean grain, in particular, has among the highest protein concentration of the major grain crops (Sinclair and deWitt 1975) with a nitrogen concentration of grain of approximately 6.4 g N/100 g. The proportion of nitrogen in soybean grain at maturity that is derived from nitrogen fixation ranges from 25 to 50 %, in fertile soils with large amounts of mineral nitrogen, to 80 to 94 %, in soils low in organic matter and nitrogen (Harper 1987; Mastrodomenico and Purcell 2012).
Nitrogen fixation in soybean occurs in root nodules formed by the microsymbiont, Bradyrhizobium japonicum, infecting and colonizing root cortical cells (Gage 2009). Signaling between plant and bacteria is intimately involved in the nodule-formation process (Gage 2009). Grafting studies between supernodulating soybean mutants and wild-type soybean indicate that lateral-root and nodule meristems send signals to shoots which in turn respond with an inhibitor to the roots, downregulating further nodulation (Caetano-Annolès and Gresshoff 1991; Gage 2009).
Differences in nodulation have been noted among soybean genotypes (Burias and Planchon 1990; Greder et al. 1986; King and Purcell 2001; Nicolás et al. 2006; Sinclair et al. 1991; Tanya et al. 2005). Sinclair et al. (1991) found differences in both nodule weight per plant and nodule number per plant and that both of these traits were significantly correlated with shoot dry weight (0.40 ≤ r ≤ 0.70). Importantly, nodule weight and seed yields are also positively correlated (Greder et al. 1986; Burias and Planchon 1990). From three small F3-derived populations (≤46 lines each), Greder et al. (1986) found broad-sense heritabilities for nodule weight from indigenous B. japonicum symbioses in field experiments to range from 0 to 0.66 and with heritabilities over 0.50 when combined over three locations for the three populations.
There have been only a few reports of nodule traits being genetically mapped in soybean, and all of these reports were from greenhouse experiments with a limited genome coverage (Nicolás et al. 2006; Santos et al. 2013; Tanya et al. 2005). For example, Nicolás identified QTLs for nodule weight, nodule number, and individual nodule weight in an F2 population of 160 plants, but only 45 informative markers were used in the study. Tanya et al. (2005) also evaluated nodule-related traits in 136 RILs from an F5-derived population using 85 SSR markers. Although several QTLs were identified for nodule traits by Tanya et al. (2005), the genetic map did not provide complete genome coverage and mapping methods were restricted to single marker analysis and not confirmed by other methods such as composite interval mapping. Santos et al. (2013) described QTLs for nodule number and individual nodule weight on a population of 157 F2:7-derived lines using multiple-trait composite interval mapping, although only 50 % of the genome was covered with 97 SSRs.
Our objectives were to first determine inheritance of nodule number, nodule weight, nodule size, and individual nodule weight from a segregating soybean population under field conditions. A second objective was to determine QTLs for these traits.
Materials and methods
Derivation of a population of 97 RILs
Two soybean (Glycine max) cultivars, KS4895 (Schapaugh and Dille 1998) and Jackson (Johnson 1958) were mated as a female and male, respectively. These two genotypes were chosen because previous research had determined that Jackson had fewer nodules per plant than did KS4895 and that the weight of individual nodules from Jackson were greater than those from KS4895 (Purcell et al. 2000; King and Purcell 2001). Additionally, Jackson has prolonged N2 fixation during the early stages of drought compared to most genotypes (Sall and Sinclair 1991) including KS4895 (Purcell et al. 1997; King and Purcell 2001). F2 seeds were harvested from F1 plants, and from the F2 to F3 or F2 to F5 generation, the population was advanced by the single seed descent method. All the seeds from individual F3 or F5 plants were threshed to generate a total of 97 RILs from 17 F3- and 80 F5-derived rows with a narrow range of maturity. For genetic analysis, F3- and F5- derived lines were combined, as described by Charlson et al. (2009), to increase mapping resolution.
Phenotyping of nodule traits
Ninety seven RILs were evaluated for nodule number (no. plant−1), individual nodule dry weight (mg nodule−1), individual nodule size (mm nodule−1), and total nodule dry weight (g plant−1) under irrigated conditions. A randomized complete block design (RCBD) was used for the field experiments. Field trials in 2000 (three reps), 2007 (one rep), and 2011 (two reps) were conducted under irrigated conditions at Fayetteville, AR (Lat. 36º5′4˝N, Long. 94º10′29˝W). In all 3 years a combination of F3- and F5-derived lines were used, but a different number of RILs was used each year due to availability of seeds. In 2000, 79 RILs were used; in 2007, 89 RILs were used; and in 2011, 86 RILs were used. Sowing dates were 13 June 2000, 16 June 2007, and 1 June 2011. Although the data in 2007 were from only one replication, it was included in the QTL analysis because data were collected on most RILs and it offered an additional environment for evaluation.
All entries were planted in one- or two-row plots (46 cm apart and 9.1 m long). Irrigation was initiated with a sprinkler system at an estimated soil-moisture deficit of 35 mm (Purcell et al. 2007). The soil at Fayetteville was Taloka silt loam (Mollic Albaqulf: fine, mixed, thermic) with 1.1 % organic matter and a pH of 6.6. Intact root systems from three plants for each RIL were carefully sampled from plants between the V7 and V9 development stages (Fehr and Caviness 1977). Intact roots were excavated to a depth of approximately 30 cm, washed in the field to remove soil, detached from the shoots, sealed in plastic bags, and transported to the laboratory. In the laboratory, roots and nodules were stored at 5 °C until they were washed again to remove any remaining soil. Nodules were manually detached from roots, scanned with a flat-bed scanner, dried, and weighed. Scanned images were analyzed using SigmaScan Pro (V.5.0 Systat Software Inc., USA) to determine nodule number and the longest dimension of the ellipsoid nodules (individual nodule size). Nodules were dried at 60 °C for at least 4 days and weighed. Individual nodule weight was determined as the quotient of total nodule weight and nodule number. Nodule number and nodule weight were expressed on a per plant basis.
Phenotypic data were analyzed using SAS 9.2 (SAS Institute Inc., Cary, NC, USA, 2008). A mixed or fixed model was used for analysis of variance (ANOVA), and heritability estimates, and least square means (LS means). Analysis of variance was performed on the data collected each year or combined over years. Normality was tested with LS means each year. A 5 % false-positive value was chosen as a significant criterion.
Genotyping of 97 RILs
Leaf tissue was freeze-dried in a lyophilizer (Model 18DX48SA, Botanique Preservation Equipment. Inc., Peoria, AZ, USA), ground to a fine powder with a pulverizer (Garcia Manufacturing, Visalia, CA, USA), and then DNA was extracted with the Maxwell 16™ (Promega, Madison, WI, USA).
A combination of SSR and SNP markers were used for genotyping. SSR markers were amplified using PCR (Akkaya et al. 1995), and amplicons were separated by electrophoresis in polyacrylamide gel or by using an ABI 3730 XL sequencer (Applied Biosynstems, Foster City, CA, USA). The Illumina GoldenGate Assay with the BeadStation 500G (http://www.illumina.com) was used to genotype 1,536 SNPs using the USLP 1.0 array (Hyten et al. 2010). The Illumina GenomeStudio software (Illumina, Inc., San Diego, CA, USA) was used to call SNP alleles. Additional SNPs that were excluded in USLP 1.0 markers were genotyped with a KASP (K-Bioscience, Hoddesdon Herts, UK), and these SNPs were analyzed by a LightCycler 480 (Roche Applied Science, Indianapolis, IN, USA) based on endpoint genotyping. We used the SNP number (ss#) assigned by the National Center for Biotechnology Information, dbSNP. We eliminated the first letters (ss) and four digits (1079) from the dbSNP ss# to prevent errors in MapMaker 3.0 (Lander et al. 1987) and Map Manager QTX (Manly et al. 2001).
QTL mapping
A description of the genetic map for this population has been reported in detail previously (Hwang et al. 2013). In summary, 664 polymorphic markers were used to construct the map, and using a minimum LOD value of 3.0, all sub-linkage groups were joined to give 20 chromosomes. Average distance between flanking markers was less than 20 cM although the distance between markers on some chromosomes exceeded 30 cM.
The software WinQTLCartographer 2.5.010 was used for composite interval mapping (CIM, Zeng 1994) and multiple-trait analysis (Jiang and Zeng 1995) as a single-QTL model. For CIM, the stepwise selection was used for background marker selection as co-factors in the model. An alpha value of 0.05 was used to avoid model over-fitting. The ML approach (Weller 1986) with the EM algorithm (Meng and Rubin 1993) was used for estimation of parameters in the model. A 1,000-repetition permutation (Churchill and Doerge 1994) was performed to find the genome-wide critical likelihood ratio test (LRT) value according to trait and year at an overall α value of 0.05. A window size of 1 cM was applied to control background marker effects and produce a precise LOD profile.
The main purpose of multiple-trait analysis was to evaluate the significance of the interaction between a QTL and years. A major difference between CIM and multiple-trait analysis was the permutation step calculating the threshold LRT. The same three traits in 2000, 2007, and 2011 were jointly and independently randomized with 1,000 repetitions to select critical threshold LRT. Consequently, four types of threshold values (i.e., 2000-, 2007-, 2011, and joint-trait) were obtained. If the LOD value of joint-trait was greater than the threshold value, the null hypothesis (Ho: QTL × Year = 0) was rejected.
Multiple interval mapping (MIM) (Kao et al. 1999) was applied in QTL Network (Yang et al. 2007, 2008) as a multiple-QTL model. First, significant marker intervals were identified by the marker-pair-selection method (Piepho and Gauch 2001). Then, a one-dimension (1D) genome scan was executed to identify QTL controlling marker intervals. For the next step, all possible epistasis between marker intervals were identified. In a two-dimension genome scan, all possible combinations of two loci were tested to determine if they had a significant effect on each nodule trait, regardless of whether or not loci were in a QTL region (Yang et al. 2007, 2008). The statistical significance of all tests was executed with an F-test. The Bayesian estimation with Gibbs sampling (Wang 1994) was used in the MIM model. The permutation test (1,000 times) was only performed in coefficient numbers of new treatment effect terms at each sequential model at an overall α value of 0.05. The QTL Network produced an F-statistic profile instead of a LOD score. Closely localized QTLs were considered as one QTL in CIM when their 95 % LOD intervals overlapped and in MIM when their 95 % confidence intervals overlapped.
Results
Phenotype data
The data for nodule number, individual nodule dry weight (individual weight), individual nodule size (size), and total nodule dry weight (total weight) in 2000 and 2007 field trials were collected at V8 to V9 (44–46 days after planting), whereas the phenotype data in 2011 was obtained at V7 to V8 (36 days after planting). The ANOVA in 2000 and 2011 indicated significant (P ≤ 0.05) differences for nodule number, and the ANOVA in 2000 indicated significant differences for nodule size (Table 1). For 2000 and 2011, the ANOVA was not significant for individual nodule weight and total nodule dry weight. An ANOVA over years for nodule number and total nodule weight indicated that the main effects of RIL and Year and their interaction were significant (P ≤ 0.05, Table 1). For individual nodule weight and size, the RIL effect was significant, but the Year and RIL × Year effects were not significant for either trait. Although the RIL × Year interaction for individual nodule weight and size were not significant, we conducted QTL analysis for all nodule traits by years since the RIL × Year interaction was significant for the other traits (nodule number and total nodule weight). In addition, LS means over years were determined for individual nodule weight and nodule size for QTL analysis.
We tested differences between the means of the parents in 2000 and 2011 (Table 2, there were no parental data in 2007). Parental differences were not significant for any of the nodule traits. For all traits in both 2000 and 2011, however, the range of the RILs exceeded that of the parents, indicating the possibility of transgressive segregation.
The population means for nodule number, individual weight, size, and total weight averaged across 3 years were 78 (no. plant−1), 2.31 (mg nodule−1), 2.44 (mm nodule−1), and 0.19 (g plant−1) (Table 2). Averaged means for the parents across the 2 years were 61 (no. plant−1), 2.26 (mg nodule−1), 2.49 (mm nodule−1), and 0.14 (g plant−1) for nodule number, individual weight, size, and total weight, respectively. Population means were close to mid-parent means except for nodule number.
Since the CIM model for QTL analysis generally assumes that traits follow a normal distribution, we tested the hypothesis that residuals follow a normal distribution and are independent for each trait each year (Table 2). When progeny mean values were used to test for normality, all nodule traits, except for nodule number plant−1 in 2011, followed a normal distribution. Although statistically not a normal distribution, the distribution of nodule number plant−1 in 2011 did not deviate greatly from a normal distribution (Fig. 1). Therefore we proceeded with the QTL analysis for nodule number plant−1 along with the other traits.

Frequency distribution of nodule number in the KS4895 × Jackson population in 2011. The solid line represents the normal curve
The broad-sense heritability for nodule traits was estimated on a progeny-mean basis over 3 years (Knapp et al. 1985). Heritability for nodule number plant−1, individual nodule weight, nodule size, and total weight plant−1 across years was 0.41, 0.42, 0.45, and 0.27, respectively (Table 1). Since the variances of mean square of error (MSE) and the RIL × Year interaction were greater than the variance of RILs, the estimated heritability for nodule traits had low values, indicating that environmental conditions (rather than additive effects) played a major role in determining phenotypes. Tanya et al. (2005) reported that heritability in a greenhouse study for nodule number (no. plant−1) and total weight (g plant−1) was 0.78 and 0.55, respectively. Similarly, Santos et al. (2013) found that heritability for nodule number (no. plant−1), individual nodule weight (mg nodule−1), and total weight (mg plant−1) was 0.33, 0.27, and 0.33, respectively.
Nodule number had a strong positive correlation with total nodule weight with correlation coefficients ranging from 0.57 to 0.74 (Table 3). Tanya et al. (2005) reported that the correlation between nodule number and total weight (g plant−1) was 0.58 in soybean. Santos et al. (2013) reported that the correlation between nodule number and total weight (mg plant−1) was 0.64 in soybean. In pea, Bourion et al. (2010) found that the correlation between nodule number and total weight (g plant−1) ranged from 0.74 to 0.77 at vegetative and flowering stages and 0.86 at beginning of seed filling. Tominaga et al. (2012) demonstrated that the correlation between nodule number and total weight (mg plant−1) was 0.52 in a Lotus japonicus population. When nodule number was regressed on total weight using average values of RILs across years, the linear regression coefficient was positive with a nodule number increase of 1.0 (no. plant−1) increasing total weight by 0.0022 g plant−1 (Fig. 2). Differences in nodule number accounted for approximately 49 % of the total weight variation in the linear regression.

Total nodule dry weight versus nodule number in a KS4895 × Jackson population. Values are from individual RILs averaged across years
Individual nodule weight and size had a positive relationship each year. The correlation between individual nodule weight and size ranged from 0.76 to 0.84 among years (Table 3). Similarly, there was a consistent positive correlation between individual weight and total weight with the correlation coefficient ranging from 0.33 to 0.64. Santos et al. (2013) reported that the correlation between individual weight and total weight was 0.24. In 2007 and 2011, there was also a significant positive correlation between total weight and size (r = 0.28 and 0.55, respectively).
Previous research has reported that plants with few nodules tend to have large nodules and vice versa (King and Purcell 2001; Purcell et al. 1997; Singleton and Stockinger 1983). In 2000 and 2007, there was a negative correlation between nodule number and size (Table 3). Likewise, in 2000, there was a negative correlation between nodule number and individual nodule weight. Similarly, Santos et al. (2013) found a negative correlation between nodule number and individual nodule weight (r = −0.40).
Genetic mapping and QTL analysis
Nodule number plant−1 QTL
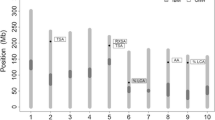
Eight QTLs for nodule number plant−1 were identified with CIM across the 3 years (Table 4; Fig. 3). These QTLs were located on Gm01 (1), Gm06 (2), Gm08 (1), Gm09 (1), Gm15 (1), and Gm20 (2). Two QTLs positioned at 80.8 (identified in 2000) and 90.3 cM (identified in 2007) on Gm06 may be the same QTL considering their overlapping 95 % LOD intervals. Additive effects ranged from 5.0 to 15.5 (no. plant−1) and the R2 values ranged from 0.14 to 0.20. The marker, BARC-047715-10388, was closest to the QTL on Gm06 that had the highest additive effect (15.5 no. plant−1). This marker also accounted for 20 % of the variation of nodule number plant−1. The KS4895 allele on all LGs contributed to an increase in nodule number plant−1. In previous research in soybean, Tanya et al. (2005) found a QTL on Gm20 associated with nodule number plant−1 close to the marker Satt440. The marker Satt440 was very tightly linked with BARC-062771-18047 (0.48 cM distance on consensus map), which was identified as a QTL for nodule number plant−1 in our population. There was no significant QTL × Year interaction for nodule number plant−1 when the multiple-trait analysis based on CIM model was applied.

Positions of QTLs for nodule traits based upon CIM in a KS4895 × Jackson population. The vertical lines on each side of QTLs represent 95 % confidence intervals. Asterisk (*) indicates mean positions of QTLs identified by MIM. Two QTLs with asterisk on Gm01 and Gm06 were identified with both models
Two QTLs for nodule number plant−1 were identified with MIM and were located on Gm06 and Gm20 (Table 4; Fig. 3). One QTL (87.3 cM) on Gm06 was very closely localized with the QTL (89.3 cM) identified by CIM in 2007. Both QTLs had the same marker, BARC-047715-10388, as a nearest marker, and these QTLs had overlapping 95 % LOD intervals in CIM and C.I. in MIM. Additive effects based on MIM for the QTLs on Gm06 and Gm20 were 8.2 and 5.2 (no. plant−1), respectively. These two QTLs had a significant QTL × Year interaction effects, which were 8.0 (no. plant−1, P < 0.001) on Gm06 and 6.6 (no. plant−1, P = 0.0012) on Gm20 in 2007. In addition, there was an interaction from a pair of loci that produced a significant effect, although these loci were not identified as QTLs in either CIM or MIM (data was not shown). One locus on Gm04 and one locus on Gm19 had a significant interaction effect of 3.8 (no. plant−1, P = 0.0061). The markers BARC-053219-11764 (ss107923464) and BARC-061089-17307 (ss107928331) were the nearest markers for these two loci.
Individual nodule weight QTL
Six QTLs for individual nodule weight were detected with CIM analysis by year (Table 4; Fig. 3). QTLs were located on Gm07 (2), Gm17 (1), Gm18 (1), and Gm19 (2). Additive effects ranged from 0.12 to 0.32 (mg nodule−1). The marker, BARC-060587-16731, was closest to the QTL on Gm19, had the highest additive effect (0.32 mg nodule−1), and accounted for 38 % of the variation. The Jackson alleles on Gm17, Gm18, and Gm19 and the KS4895 alleles on Gm07 contributed to an increase in individual nodule weight.
Another QTL for individual nodule weight was identified with MIM on Gm07 at 43.1 cM (Table 4). This QTL was close to the two QTL identified on Gm07 in 2007 using CIM. The additive effect for this QTL identified on Gm07 by MIM was 0.13 (mg nodule−1), and the KS4895 allele contributed to an increase in individual nodule weight as did the KS4895 allele for both QTL on GM07 identified by CIM analysis. In both CIM and MIM, significant QTL × Year or other interactions were not detected for individual nodule weight.
Because the RIL by year interaction term was not significant for individual nodule weight, but the main effect of RIL was significant, we also evaluated possible QTLs for individual nodule weight using the LS means for RILs over years. The CIM analysis using LS Mean values for RILs over years identified two unique QTLs that were not found in the QTL analysis conducted by year (Table 4). These QTLs were located on Gm01 and Gm05 and accounted for 15 and 12 % of the phenotypic variation, respectively. For both of these QTLs, Jackson alleles conditioned an increase in individual nodule weight.
Nodule size QTL
Seven QTLs for nodule size were identified with CIM analysis by year (Table 5; Fig. 3). QTLs were located on Gm01 (1), Gm04 (1), Gm05 (1), Gm06 (1), Gm15 (1), and Gm19 (2). Additive effects ranged from 0.06 to 0.10 (mm). The marker Satt713, closest to the QTL on Gm04, had the highest additive effect, whereas the marker BARC-060587-16731 on Gm19 accounted for the highest phenotypic variance (27.2 %). The Jackson alleles on Gm01, Gm06, Gm15, and Gm19 and the KS4895 alleles on Gm04 and Gm05 contributed to an increase of size. There was no QTL detected using MIM. In both CIM and MIM, there was no significant QTL × Year or other interactions.
It appeared that four QTLs for nodule size on Gm06 (1), Gm15 (1), and Gm19 (2) had pleiotrophic effects with QTLs for nodule number plant−1 and individual nodule weight considering their overlapping 95 % LOD intervals and strong phenotypic correlations (Table 3; Fig. 3). In all cases for these QTLs, the additive effect for a pleiotrophic QTL pair was in the expected direction based upon correlations between traits. For example, nodule number and nodule size were negatively associated in 2000 (Table 3) and the allele contributing the additive effect at the pleiotrophic QTL on Gm06 in 2000 for nodule number (80.8 cM) and nodule size (83.3 cM) had opposing effects. Similarly, nodule size and individual nodule weight were positively associated (Table 3) and both pleiotrophic QTLs on Gm19 in 2011 had positive additive effects from the Jackson alleles for nodule size and individual nodule weight.
As described for individual nodule weight, we also conducted a QTL analysis for nodule size using LS Mean values for RILs over years. This analysis identified a unique QTL on Gm11 that accounted for 12 % of the variation and that was not identified in the analysis conducted by year (Table 5).
Nodule weight plant−1 QTL
Five QTLs for total nodule weight plant−1 were detected with CIM (Table 5; Fig. 3). QTLs were located on Gm01 (1), Gm06 (1), Gm15 (2), and Gm18 (1) with additive effects ranging from 0.01 to 0.05 g plant−1. The marker BARC-007726-00090, closest to the QTL on Gm01, had the highest additive effect and accounted for the highest phenotypic variance (20 %). The KS4895 alleles on all chromosomes contributed to an increase in total nodule weight. Significant QTL × Year interactions for total nodule weight were not identified when the multiple-trait analysis was applied. One QTL on Gm01 for total weight seemed to have a pleiotrophic effect with a QTL (77.6 cM) identified for nodule number. The KS4895 QTL at this location appears to confer a greater nodule number resulting in greater total weight, which agrees with the expected response based upon the phenotypic correlation between nodule number and total nodule weight (Table 3).
One QTL for total nodule weight plant−1 was identified with MIM (Table 5). This QTL (79.5 cM) on Gm01 was closely located with a QTL (80.6 cM) for total nodule weight plant−1 identified by CIM in 2007. Both QTLs had the same marker, BARC-007726-00090, as a nearest marker. The additive effect for this QTL was 0.02 (g plant−1) with the KS4895 allele contributing to increased total nodule weight plant−1. The QTL had a significant QTL × Year interaction effects, which were 0.03 (g plant−1, P < 0.001) in 2007 and 0.02 (g plant−1, P = 0.039) in 2011.
Neighboring traits and QTL
We also searched SoyBase (http://soybase.org/) for other traits with QTL positions similar to the positions that we found for nodule number plant−1, individual nodule weight, nodule size, and total nodule weight plant−1. A complete list of those QTLs is shown in Table 6. We have described below those QTLs for traits that may have some functional relationship with nodulation or nitrogen fixation (e.g., yield, protein, nitrogen). QTLs for nodule number plant−1 were close to QTLs for yield or seed weight (Orf et al. 1999; Kabelka et al. 2004; Du et al. 2009), and seed protein or amino acid-related traits (Reinprecht et al. 2006; Csanadi et al. 2001; Panthee et al. 2004, 2006; Primomo et al. 2005). QTLs for individual nodule weight were located near QTLs for seed protein (Reinprecht et al. 2006; Panthee et al. 2004, 2006), and shoot N (Hwang et al. 2013). QTLs for nodule size were located near QTLs for seed yield (Du et al. 2009; Kabelka et al. 2004), and seed protein or amino-acid related traits (Kabelka et al. 2004; Panthee et al. 2006). For total nodule weight plant−1, QTLs were located near reported QTLs for glycitein content (Primomo et al. 2005), seed protein, yield (Reinprecht et al. 2006), and seed weight (Hyten et al. 2004).
Discussion
Statistical power for QTL detection
All nodule traits we measured had low to moderate heritability ranging from 0.27 to 0.45 when determined over the multiple environments. For nodule number plant−1 and total nodule weight plant−1, there were highly significant year by RIL interactions. Therefore, QTL analysis for nodule number plant−1 and total nodule weight was conducted separately for each year. Conversely, for both individual nodule weight and nodule size, the year by RIL interaction was not significant but the main effect of RIL was highly significant, and, therefore, for these traits we conducted QTL analysis by year as well as over years.
Surprisingly, none of the QTLs identified in individual years for either individual nodule weight or nodule size were detected in the analysis conducted over years. In fact, QTLs identified over years for individual nodule weight and nodule size were on completely different chromosomes from those identified in individual years. Phenotypic correlations of individual nodule weight or nodule size had relatively poor association among individual years (0.16 ≤ r ≤ 0.39, data not shown), but correlations of these variables for individual years with LS means over years were much higher (0.53 ≤ r ≤ 0.83). Because of the low heritability of individual nodule weight and nodule size and because none of the QTLs found in the analysis over years matched those found by individual years, we question the reliability of the QTLs identified by the analysis of LS means over years. Nonetheless, we include these results as a resource for future work mapping nodule traits in other populations or environments.
We investigated the statistical power of detected QTLs for nodule traits with the qtlDesign library in R (Broman et al. 2003; Sen et al. 2007), which provided a simulation approach with a single QTL model. The minimum detectable QTL effect on individual nodule weight was 0.17 (mg nodule−1) and the R 2 value of a QTL exhibiting this minimum QTL effect was 18.8 % using parameters to be considered from a KS4859 × Jackson population study. The data from a KS4895 × Jackson population indicated R 2 values ranged from 12 to 38 % and QTL effects ranged from 0.12 to 0.32 (mg nodule−1, Table 4). One QTL on Gm18 had a R 2 value of 12 % and an additive effect of 0.12 mg nodule−1, and this QTL had statistical power less than 0.8. In the case of nodule number plant−1, nodule size, and total nodule weight plant−1, we found that the R 2 value and additive effects from the simulation had similar ranges as those from KS4895 × Jackson population study.
Separation of linked QTL
The genomic position of some QTLs were in close proximity to the position of other QTLs, particularly on chromosomes 19 and 20. To separate adjacent QTLs, we used 5 cM as the minimum distance and 1 LOD as the minimum value to distinguish the top and bottom of a QTL peak. For example, two QTLs for nodule number on Gm20 in 2007 were detected by CIM with close map positions of 74.9 cM and 84.3 cM (Table 4). Based upon our criteria for LOD intervals, these adjacent QTLs appeared to be distinct. However, the total phenotypic variance explained by four QTLs in 2007 was high (69 %) considering the broad-sense heritability of nodule number (0.32–0.41, Table 1). Further evaluation with MIM indicated that these two QTLs could be considered as one QTL (73.9 cM). Therefore, there is a possibility that there is one QTL in this region rather than two.
In case of the two QTLs on Gm19 (99.7 and 105.2 cM) for individual nodule weight, the total phenotypic variance explained by three QTLs in 2011 was also high (82 %) for a trait with low broad-sense heritability (0.13–0.42, Table 1). MIM failed to identify any QTL in this region, and it may be that there is only one QTL in this area. In this same region on Gm19, there were two QTL for nodule size that were also close together (97.5 and 108.2 cM, Table 5) but whose LOD intervals did not overlap. Given the relatively low broad-sense heritability for nodule size (0.23–0.45, Table 1), only one of these may be an actual QTL. Determination if these QTLs on Gm19 and Gm20 are truly separate QTLs will likely require careful phenotypic evaluation in a larger population.
Association of nodule traits with other traits
Several of the QTLs identified for nodule traits were previously associated with yield or yield components such as seed weight and seed oil or protein content (Table 6). Previous studies reported that seed protein, oil, and yield have been consistently reported as having high phenotypic or genotypic correlations, and most of the QTLs for these traits were pleiotrophically co-localized (Chung et al. 2003; Hanson et al. 1961; Helms and Orf 1998; Thorne and Fehr 1970; Wilcox and Cavins 1995). Additional QTLs for nodule traits were related with plant growth (photoperiod sensitivity, beginning pod, maturity date, flowering date, height and lodging) and biotic-stress against fungal and insect pests. The population study by Tanya et al. (2005) showed that the correlations coefficients between N2 fixation activity from acetylene reduction assay (ARA) and nodule number plant−1 or nodule fresh or dry weight plant−1 were 0.44, 0.74, and 0.70, respectively. Pazdernik et al. (1996) reported that the correlation coefficients between ARA and nodule number plant−1 or fresh weight plant−1 were 0.45 and 0.86, respectively. Additionally, Greder et al. (1986) and Burias and Planchon (1990) reported positive association between nodule weight plant−1 and yield. These results indirectly imply that QTLs for nodule traits can contribute to increased N2 fixation and yield.
With two exceptions, previously reported QTLs for total nodule weight, individual nodule weight, nodule number, and nodule size in soybean (Nicolás et al. 2006; Tanya et al. 2005; Santos et al. 2013) did not co-localize with QTLs found in the current research. The QTL for nodule number plant−1 on Gm20 (82.9 cM) in 2007 (Table 4) was close to the QTL for nodule number plant−1 found by Tanya et al. (2005). Additionally, the QTL for nodule size that we found on Gm11 using the combined data over years (Table 5) was close to a QTL that Santos et al. (2013) reported that had pleiotrophic effects on nodule number plant−1, individual nodule weight, and shoot dry weight.
There are several reasons why other QTLs we identified were not identified in previous research. Genetic background of material from the U.S. (our research), Korea (Tanya et al. 2005), and Brazil (Nicolás et al. 2006; Santos et al. 2013) was likely different and may have had unique loci associated with nodule traits. Also, earlier reports used very limited genetic mapping information (45 markers by Nicolás et al. 2006, and 85 markers by Tanya et al. 2005) to identify QTL compared to the current research (664 markers), which may have precluded discovery of QTLs in past research. Additionally, our research indicated that environment may influence the expression of nodule traits, and environmental effects on nodule traits from plants grown in the field (current research) versus a greenhouse (Nicolás et al. 2006; Tanya et al. 2005; Santos et al. 2013) are likely quite large.
Possible mechanisms or factors affecting nodule traits
In soybean, the Rj/rj loci have been associated with nodulation responses since the 1950s (Williams and Lynch 1954). Some loci have evolved naturally while others were identified by artificial mutagenesis, allelism and complementation tests, and genetic mapping. The Rj 7 allele confers autoregulation of nodulation in the host plant (Caetano-Annolès and Gresshoff 1991; Searle et al. 2003; Schnabel et al. 2005). Disruption of nodule autoregulation with the rj 7 allele results in excessive or hypernodulation (Caetano-Annolès and Gresshoff 1991), but there was no association of nodule number with rj 7 (Gm12) in our population.
The three recessive alleles, rj 1 (nod49), rj 5 (nod139-1), and rj 6 (nod139-2) result in a non-nodulation phenotype (Williams and Lynch 1954; Indrasumunar et al. 2010, 2011) and were investigated to determine if individual or total nodule weight were associated with these loci in our population. There were no QTLs associated with rj 1 (Gm02), but two of the markers associated with rj 6, Satt408 and Satt071 (Indrasumunar et al. 2010), mapped closely to a QTL we found on Gm01 (80.6 cM) in 2007 for total nodule weight based upon CIM (Table 5). Interestingly, this same QTL for total nodule weight on Gm01 (80.6 cM) had an overlapping LOD interval with a QTL for nodule number that we identified in 2007 (77.6 cM).
The QTL we identified for nodule size on Gm11 (0.0 cM) by CIM using combined data over years (Table 5) was associated with rj 5, which was cloned as a NOD factor receptor gene (GmNFR5α) (Indrasumunar et al. 2010). Santos et al. (2013) reported that a QTL in this region, identified by multiple-trait CIM analysis, apparently had pleiotrophic effects on nodule number, individual nodule weight, and shoot dry weight.
Conclusion
We identified QTLs for nodule number, average nodule weight, nodule size, and total nodule weight under irrigated field conditions. These results represent the first QTL information on nodule traits in soybean from field experiments using a dense and complete linkage map. Because individual nodule weight and total nodule weight are closely associated with N2 fixation (Burias and Planchon 1990; Greder et al. 1986; Pazdernik et al. 1996; Tanya et al. 2005), we expect that our population study for nodule traits will be helpful in selecting genotypes with increased capacity for N2 fixation.
Abbreviations
- C.I.:
-
Confidence interval
- CIM:
-
Composite interval mapping
- LG:
-
Linkage group
- LS:
-
Least square means
- LOD:
-
Logarithm of odds
- LRT:
-
Likelihood ratio test
- MIM:
-
Multiple interval mapping
- QTL:
-
Quantitative trait loci
- RIL:
-
Recombinant inbred line
- SSR:
-
Simple sequence repeat
- SNP:
-
Single nucleotide polymorphism
References
Akkaya MS, Shoemaker RC, Specht JE, Bhagwat AA, Cregan PB (1995) Integration of simple sequence repeat DNA markers into a soybean linkage map. Crop Sci 35:1439–1445
Arahana VS, Graef GL, Specht JE, Steadman JR, Eskridge KM (2001) Identification of QTLs for Resistance to Sclerotinia sclerotiorum in Soybean. Crop Sci 41:180–188
Bourion V, Rizvi SMH, Fournier S, Larambergue HD, Galmiche F, Marget P, Duc G, Burstin J (2010) Genetic dissection of nitrogen nutrition in pea through a QTL approach of root, nodule, and shoot variability. Theor Appl Genet 121:71–86
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Burias N, Planchon C (1990) Increasing soybean productivity through selection for nitrogen fixation. Agron J 82:1031–1034
Caetano-Annolès G, Gresshoff PM (1991) Plant genetic control of nodulation. Ann Rev Microbiol 45:345–382
Charlson DV, Bhatnagar S, King CA, Ray JD, Sneller CH, Carter TE Jr, Purcell LC (2009) Polygenic inheritance of canopy wilting in soybean [Glycine max (L.) Merr.]. Theor Appl Genet 119(4):587–594
Chung J, Babka HL, Graef GL, Staswick PE, Lee DJ, Cregan PB, Shoemaker RC, Specht JE (2003) The seed protein, oil, and yield QTL on soybean linkage group I. Crop Sci 43:1053–1067
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:967–971
Csanadi G, Vollmann J, Stift G, Lelley T (2001) Seed quality QTLs identified in a molecular map of early maturing soybean. Theor Appl Genet 103:912–919
Du W, Wang M, Fu S, Yu D (2009) Mapping WTLs for seed yield and drought susceptiblity index in soybean (Glycine max L.) across different environments. J Genet Genomics 36:721–731
Fehr WR, Caviness CE (1977) Stages of soybean development. Coop Ext Serv Spec Rep 80. Iowa State University, Ames
Gage DJ (2009) Nodule development in legumes. In: Emerich DW, Krishnan HB (eds) Nitrogen fixation in crop production. ASA, CSSA, SSSA, Madison, pp 1–24
Greder RR, Orf JH, Lambert JW (1986) Heritabilities and associations of nodule mass and recovery of Bradyrhizobium japonicum serogroup USDA 110 in soybean. Crop Sci 26:33–37
Hanson WD, Leffel RC, Howell RW (1961) Genetic analysis of energy production in the soybean. Crop Sci 1:121–126
Harper JE (1987) Nitrogen metabolism. In: Wilcox JR (ed) Soybeans: Improvement, production, and uses. 2nd edn. Agron Monogr 16, ASA, CSSA, SSSA, Madison, WI, p 497–533
Helms TC, Orf JH (1998) Protein, oil and yield in soybean lines selected for increased protein. Crop Sci 38:707–711
Hwang S, King CA, Davies MK, Ray JD, Cregan PB, Purcell LC (2013) QTL analysis of shoot ureide and nitrogen concentrations in soybean. Crop Sci (in press)
Hyten DL, Pantalone VR, Sams CE, Saxton AM, Landau-Ellis D, Stefaniak TR, Schmidt ME (2004) Seed quality QTL in a prominent soybean population. Theor Appl Genet 109:552–561
Hyten DL, Choi IY, Song Q, Specht JE, Carter TE Jr, Shoemaker RC, Hwang EY, Matukumalli LK, Cregan PB (2010) A high density integrated genetic linkage map of soybean and the development of a 1536 universal soy linkage panel for quantitative trait locus mapping. Crop Sci 50:1–9
Indrasumunar A, Kereszt A, Searle I, Miyagi M, Li D, Nguyen CDT, Men A, Carroll BJ, Gresshoff PM (2010) Inactivation of duplicated Nod factor receptor 5 (NFR5) genes in recessive loss-of-function non-nodulation mutants of allotetraploid soybean (Glycine max L. Merr.). Plant Cell Physiol 51:201–214
Indrasumunar A, Searle I, Lin MH, Kereszt A, Men A, Carroll BJ, Gresshoff PM (2011) Nodulation factor receptor kinase 1α controls nodule organ number in soybean (Glycine max L. Merr). Plant J 65:39–50
Jiang C, Zeng Z-B (1995) Multiple trait analysis and genetic mapping for quantitative trait loci. Genetics 136:1447–1455
Johnson HW (1958) Registration of soybean varieties, VII. Agron J 11:659–660
Kabelka EA, Diers BW, Fehr WR, LeRoy AR, Baianu IC, You T, Neece DJ, Nelson RL (2004) Putative alleles for increased yield from soybean plant introductions. Crop Sci 44:784–791
Kao C-H, Zeng Z-B, Teasdale RD (1999) Multiple interval mapping for quantitative trait loci. Genetics 152:1203–1216
Kim H, Kang S, Cho J, Choung M, Suh D (2005) Quantitative trait loci associated with oligosaccharide and sucrose contents in soybean (Glycine max L.). J Plant Biol 48:106–112
King CA, Purcell LC (2001) Soybean nodule size and relationship to nitrogen fixation response to water deficit. Crop Sci 41:1099–1107
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Komatsu K, Okuda S, Takahashi M, Matsunaga R, Nakazawa Y (2005) QTL mapping of antibiosis resistance to common cutworm (Spodoptera litura Fabricius) in soybean. Crop Sci 45:2044–2048
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) Mapmaker an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Manly KF, Cudmore RH Jr, Meer JM (2001) Map Manager QTX, cross-platform program software for genetic mapping. Mamm Genome 12:930–932
Mastrodomenico AT, Purcell LC (2012) Soybean nitrogen fixation and nitrogen remobilization during reproductive development. Crop Sci 52:1281–1289
Meng X-L, Rubin DB (1993) Maximum likelihood estimation via the ECM algorithm: a general framework. Biometrika 80:267–278
Narvel JM, Walker DR, Rector BG, All JN, Parrott WA, Boerma HR (2001) A retrospective DNA marker assessment of the development of insect resistant soybean. Crop Sci 41:1931–1939
Nicolás MF, Hungria M, Arias CAA (2006) Identification of quantitative trait loci controlling nodulation and shoot mass in progenies from two Brazillian soybean cultivars. Field Crops Res 95:355–366
Orf JH, Chase K, Jarvik T, Mansur LM, Cregan PB, Adler FR, Lark KG (1999) Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Sci 39:1642–1651
Panthee DR, Kwanyuen P, Sams CE, West DR, Saxton AM, Pantalone VR (2004) Quantitative trait loci for B-conglycinin (7S) and glycinin (11S) fractions of soybean storage protein. J Am Oil Chem Soc 81:1005–1012
Panthee DR, Pantalone VR, Saxton AM, West DR, Sams CE (2006) Genomic regions associated with amino acid composition in soybean. Mol Breed 17:79–89
Pazdernik DL, Graham PH, Vance CP, Orf JH (1996) Host genetic variation in the early nodulation and dinitrogen fixation of soybean. Crop Sci 36:1102–1107
Piepho HP, Gauch HG (2001) Marker pair selection for mapping quantitative trait loci. Genetics 157:433–444
Primomo V, Poysa V, Ablett G, Jackson C, Gijzen M, Rajcan I (2005) Mapping QTL for individual and total isoflavone content in soybean seeds. Crop Sci 45:2464–2545
Purcell LC (2009) Physiological responses of N2 fixation to drought and selecting genotypes for improved N2 fixation. In: Emerich DW, Krishnan HB (eds) Nitrogen fixation in crop production. ASA, CSSA, SSSA, Madison, pp 211–238
Purcell LC, deSilva M, King CA, Kim WH (1997) Biomass accumulation and allocation in soybean associated with genotypic differences in tolerance of nitrogen fixation to water deficits. Plant Soil 196:101–113
Purcell LC, King CA, Ball RA (2000) Soybean cultivar differences in ureides, drought tolerant nitrogen fixation, and manganese nutrition. Crop Sci 40:1062–1070
Purcell LC, Edwards JT, Brye KR (2007) Soybean yield and biomass responses to cumulative transpiration: questioning widely held beliefs. Field Crops Res 101:10–18
Qi Z, Wu Q, Han X, Sun Y, Du X, Liu C, Jiang H, Hu G, Chen Q (2011) Soybean oil content QTL mapping and integrating with meta-analysis method for mining genes. Euphytica 179:499–514
Reinprecht Y, Poysa V, Yu K, Rajcan I, Ablett G, Pauls K (2006) Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome 49:1510–1527
Sall K, Sinclair TR (1991) Soybean genotypic differences in sensitivity of symbiotic nitrogen fixation to soil dehydration. Plant Soil 133:31–37
Santos MA, Geraldi IO, Garcia AAF, Bortolatti N, Schiavon A, Hungria M (2013) Mapping of QTLs associated with biological nitrogen fixation traits in soybean. Hereditas 150:17–25
Schapaugh WT, Dille RE (1998) Registration of ‘KS4895′ soybean. Crop Sci 38:892
Schnabel E, Journet EP, de Carvalho-Niebel F, Duc G, Frugoli J (2005) The Medicago truncatula SUNN gene encodes a CLV1-like leucine-rich repeat receptor kinase that regulates nodule number and root length. Plant Mol Biol 58:809–822
Searle IR, Men AE, Laniya TS, Buzas DM, Iturbe-Ormaetxe I, Carroll BJ, Gresshoff PM (2003) Long-distance signaling in nodulation directed by a CLAVATA1-like receptor kinase. Science 299:109–112
Sen S, Satagopan JM, Broman KW, Churchill GA (2007) R/qtldesign: inbred line cross experimental design. Mamm Genome 18:87–93
Sinclair TR, deWitt C (1975) Photosynthate and nitrogen requirements for seed production by various crops. Science 189:565–567
Sinclair TR, Soffes AR, Hinson K, Albrecht SL, Pfahler PL (1991) Genotypic variation in soybean nodule number and weight. Crop Sci 31:301–304
Singleton PW, Stockinger KR (1983) Compensation against ineffective nodulation in soybean. Crop Sci 23:69–72
Tanya P, Srinives P, Toojinda T, Vanavichit A, Lee SH (2005) Identification of SSR markers associated with N2-fixation component in soybean [Glycine max (L.) Merr.]. Korean J Genetics 27:351–359
Tasma IM, Lorenzen LL, Green DE, Shoemaker RC (2001) Mapping genetic loci for flowering time, maturity, and photoperiod insensitivity in soybean. Mol Breed 8(1):25–35
Terry LI, Chase K, Jarvik T, Orf JH, Mansur LM, Lark KG (2000) Soybean quantitative trait loci for resistance to insects. Crop Sci 40(2):375–382
Thorne JC, Fehr WR (1970) Incorporation of high-protein, exotic germplasm into soybean populations by 2- and 3-way crosses. Crop Sci 10:652–655
Tischner T, Allphin L, Chase K, Orf JH, Lark KG (2003) Genetics of seed abortion and reproductive traits in soybean. Crop Sci 43:464–473
Tominaga A, Gondo T, Akashi R, Zheng S-H, Arima S, Suzuki A (2012) Quatitative trait locus analysis of symbiotic nitrogen fixation activity in the model legume Lotus japonicus. J Plant Res 125:395–406
Wang CS (1994) Bayesian analysis of mixed linear models via Gibbs sampling with an application to litter size in Iberian pigs. Genet Sel Evol 26:91–115
Weller JI (1986) Maximum likelihood techniques for the mapping and analysis of quantitative trait loci with the aid of genetic markers. Biometrics 42:627–640
Wilcox JR, Cavins JF (1995) Backcrossing high seed protein to a soybean cultivar. Crop Sci 35:1036–1041
Williams LF, Lynch DL (1954) Inheritance of a non-nodulation character in the soybean. Agron J 46:28–29
Yang J, Zhu J, Williams RW (2007) Mapping the genetic architecture of complex traits in experimental populations. Bioinformatics 23:1527–1536
Yang J, Hu C, Hu H, Yu R, Xia Z, Ye X, Zhu J (2008) QTL Network: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24:721–723
Zeng Z-B (1994) Precise mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
The authors gratefully acknowledge partial financial support for this research from the United Soybean Board and from the United States Department of Agriculture-Agriculture Research Service (USDA-ARS) project number 6402-21220-012-00D. Editorial assistance from Penny McGee is greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
Mention of a trademark or proprietary product does not constitute a guarantee or warranty of the product by the US Department of Agriculture and does not imply approval or the exclusion of other products that may also be suitable. USDA is an equal opportunity provider and employer.
Rights and permissions
About this article
Cite this article
Hwang, S., Ray, J.D., Cregan, P.B. et al. Genetics and mapping of quantitative traits for nodule number, weight, and size in soybean (Glycine max L.[Merr.]). Euphytica 195, 419–434 (2014). https://doi.org/10.1007/s10681-013-1005-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-013-1005-0