
Abstract
Grain moisture in maize at harvest depends on the grain drying rate (GDR) after physiological maturity. The maize plants with high GDR can reduce grain moisture rapidly, which will shorten the drying time after harvest and prevent the grain to be mildew and enhance maize quality. In this study, A total of 280 recombinant inbred lines that were derived from a cross between Ji846 (high drying rate, 1.18 % day−1) and Ye3189 (slow drying rate, 0.39 % day−1) were used to construct genetic linkage map and identify QTL underlying GDR in different environments. A genetic linkage map was constructed containing 97 SSR and 49 AFLP markers, which covered 2356.8 cM of the maize genome, with an average distance of 16.1 cM. Composite interval mapping identified 14 QTL for GDR after physiological maturity located on chromosomes 2, 3, 5, 6 and 8. The additive effects of QTL were all from Ji846. The range of phenotypic variation explained by the QTL was 5.05–16.28 %. But only two QTL (qKdr-2-1, qKdr-3-6) were identified across both locations. qKdr-2-1 positioned between the markers phi090-umc1560 on chromosome 2 explained 15.59 % of the phenotypic variance, and the other qKdr-3-6 positioned between the markers phi046-bnlg1754 on chromosome 3 explained 10.28 % of the phenotypic variance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maize (Zea Mays L.) is one of the most important grains in the world. It plays a key role in the world food economy by feeding people and being raw industrial materials. Grain moisture (GM) at harvest is of primary importance for maize production and breeding since crops with low GM at harvest can reduce the economic impact of artificial drying (Dijak et al. 1999; Sweeney et al. 1994). On average, the GM is about 30 % at harvest in the Northern spring maize growing region, the main maize production area in China. It has become the major barrier for sustainable maize development in this region (Zhang et al. 2007a). The most economical and effective way to solve this problem is to select varieties with low GM at harvest. GDR and cultivar maturity are two important factors to determine GM at harvest. GDR is defined as the rate of GM loss during the drying period, that is, from physiological maturity to grain harvest. GDR was a quantitative trait with high heritability and additive effect (Lv et al. 2006; Zhao and Wu 2002). Crane et al. (1959) reported that there were significantly difference in GDR for maize hybrids and the trait was heritable. Hallauer and Russell (1960) found that the rapid-dehydration maize had rapid GDR from physiological maturity to harvest. Troyer and Ambrose (1971) defined the rapid dehydration maize as follows: it can be harvested safely in a short time after physical maturity. High GM of maize occurs mainly in cold high-latitude areas in the world, so climate conditions is one of the reasons that influence kernel moisture content at harvest. In addition, the peasants planted the maize varieties which were not appropriate for their area and maize can not reach physiological maturity when harvested. These are the major cause for high moisture in the kernel in northern China.
A few researches showed that GDR was controlled by additive gene rather than by non-additive effect, while it has high heritability and can be stably inherited (Purdy and Crane 1967; Cai 1995). However, the genetic regulation of fast dehydration is still not clearly. GDR is a trait which was difficult to measure, So the genetic mechanism of maize natural GDR after physiological maturity should be studied to find the relative genes.
Currently, few studies have attempted to map quantitative trait loci (QTL) associated with GDR. Sala et al. (2006) identified three QTLs of GDR 1 week before harvest across two locations and 1 year, using 181 F2:3 families as the mapping population. In order to find the stably expressed QTL, the mapping and traits evaluation population with 280 RIL derived from a cross between Ji846 (high drying rate, 1.18 % day−1) and Ye3189 (slow drying rate, 0.39 % day−1) has been used for the present study. The linkage map constructed by SSR and AFLP markers and the composite interval mapping (CIM) were used to detect the number of genes, chromosome location, gene effects and mode of action related with natural GDR after physiological maturity in different environments. Our objective is to explore the stably expressed QTL in different places and years for the sake of finding tightly linked molecular markers. The results will be highly helpful in creating and selecting the new maize inbred lines with high GDR using molecular marker-assisted selection technology, reducing the GM of harvested maize in cold region, and improving of maize quality.
Materials and methods
Plant materials and field design
The mapping population, consisting of 280 F7:8 the recombinant inbred lines (RILs), was advanced by single-seed-descent (SSD) in 2003–2007 from the cross of ‘Ji846’ (developed by Jilin Academy of Agriculture Sciences, China, high GDR of 1.18 % day−1) and ‘Ye3189’ (developed by Laizhou Maize Research Institute, Shandong, China, low GDR of 0.39 % day−1) (Table 1). The parents, F1, 280 F7:8 RILs were grown at the Experimental Station of Northeast Agricultural University (Harbin, China) and the Fenghe Maize Research Institute (Shuangcheng, China) in a randomized complete block design with three replications in 2007 and 2008, respectively. The double-row plots were 4.5 m long with 0.3 m individual plant spacing, 0.7 m between rows. The plot area was 6.30 m2, and the field management at the two locations was the same in both years.
GDR after physiological maturity was evaluated by following the procedure described by Wang et al. (2001) and Jin et al. (1997). The ears were bagged before silking (50 % of plants in the row having extruded silks). Then the bagged ears were pollinated by hand. Two weeks later, the bags were removed and tested plants were tagged to keep sampling time the same after pollinated. Two ears were sampled randomly in each polt every 4 days from 45 to 70 day after pollination. A total of 150 g (W1, g) kernel were weighed for each sample and put into a mesh bag to air dry, and the air-dried kernel weight (W2, g) was marked to record. A total of 30 g air-dried kernel (W3, g) were sampled and put into an oven for 36 h at 103 ± 2 °C to a constant weight (W4, g). GM was calculated according to the formula below:
Meanwhile, randomly sampled 300 air-dried kernels were weighed and converted to 100-kernel weight with 14 % moisture. The date when the 100-kernel weight (14 % moisture) in each plot reached to their maximum was recorded as the physiological maturity period. The used formula for the average GDR was as following:
where W5 is the GDR at physiological maturity and W6 is the GDR at 10 days later for physiological maturity.
Broad-sense heritability (h2) for each trait was computed according to Knapp et al. (1985). The heritability was calculated as follows: \( {\text{h}}^{2} = \sigma^{2}_{\text{g}} /(\sigma^{2}_{\text{g}} + \sigma^{2}_{\text{ge}} /n + \sigma^{2}_{\text{e}} /nr) \)where \( \sigma^{2}_{\text{g}} \) is the genetic variance, \( \sigma^{2}_{\text{ge}} \) is genotype-by-environment interaction, \( \sigma^{2}_{\text{e}} \) is the error variance, r is the replication number, and n is the number of environments. The estimates of \( \sigma^{2}_{\text{g}} \), \( \sigma^{2}_{\text{ge}} \), and \( \sigma^{2}_{\text{e}} \) were obtained by analysis of variance (ANOVA) using the general linear model (GLM) procedure of the statistical software SAS procedure (PROC.GLM.SAS).
Linkage analysis
Total genomic DNA of F78-generation plants were isolated from leaf tissue using CTAB method (Doyle and Doyle 1990). A total of 374 pairs of SSR primers from SSR bin map (http//www.Maizegdb.org/ssr.php) and 40 pairs of AFLP primers were selected for molecular markers analysis (Senior and Manfred 1993). Genotype statistic and genetic linkage group division were based on the protocol (Yang et al. 2004). The markers that fit segregation ratio by Chi-square test were used to construct linkage map using MAPMAKER version 3.0b (Lander et al. 1987). Linkage groups were created with a LOD score of 3.0 and a recombination fraction of 0.4 by the “group” command. The ripple command was used to verify the order of markers on each chromosome. Data quality was checked by “error detection” command, and unlikely double crossovers, because of possible genotyping errors, were corrected by rechecking the data. The map distance in centimorgans (cM) was derived on the basis of the Kosambi function (Kosambi 1944).
QTL analysis
QTL, underlying mean performance of the F7:8 RILs across Harbin and Shuangcheng at 2 years, was identified using QTL Cartographer 2.5 (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm, 2008) with CIM module (Basten et al. 1996). The likelihood-ratio (LR) test statistic used was 22 ln (L0/L1), where L0/L1 is the ratio of the likelihood under the null hypothesis (there is no QTL in the interval) and the alternative hypothesis (there is a QTL in the interval). We used Model 6 of the Zmapqtl module of QTL Cartographer (Churchill and Doerge 1994; Doerge and Churchill 1996), scanning intervals of 1 cM between markers and putative QTL with a window size of 10 cM. The number of marker cofactors for background control was set via forward–backward stepwise regression. The presence of a putative QTL was declared if the log likelihood ratio (LOD) threshold was larger than 3.0. QTL were deemed to exist only at positions where a LOD score exceeded the corresponding significance threshold. The estimation of the position, genetic effects, and percentage of phenotypic variation of the QTL were performed at the significant LOD peak in the region under consideration. The QTL were named according to the method introduced by McCouch et al. (1997).
Results
Phenotypic analysis of GDR
The effects of environment, line, line × environment, and error were significant at 0.01 level. The broad heritability estimated for GDR was 77.18 % (Table 2). However, environmental influence was found on GDR, and the errors could be reduced by average values among replications and across years (Table 2). The distribution of GDR among the 280 families in 1-location/2-years and 2-locations/2-years was normal (Fig. 1). The results showed that the population constructed in this study was a random one without significant deviation, with continuous distribution of GDR, and applicable for QTL mapping.

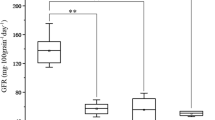
Phenotype value of field grain drying rate at two locations
The construction of genetic linkage map
A total of 97 SSR and 49 AFLP markers covered 2356.8 cM with an average interval of 16.1 cM. The intervals among 73.28 % markers were below 20 cM. In general, the basic frame of maize has been constructed by the genetic linkage map for QTL mapping. Most of the SSR markers were in agreement with those on linkage map of maize (IBM2008), and there was only slight change on loci of the chromosome for three markers, which were phi090 (Bin 2.08) on 2.07, umc1527 (Bin 3.04) on 3.05, and phi092 (Bin 4.08) on 4.11. And two markers completely changed, which were umc1018 (Bin 2.04) between 6.01 and 6.02 and phi112 (Bin 7.01) around 8.03.
QTL analysis
A total of 14 QTL for GDR were detected at two locations and were mainly distributed on chromosome 2, 3, 5, 6 and 8 (Table 3; Fig. 2). In Harbin, six QTLs (qKdr-2-1, qKdr-3-2, qKdr-3-4, qKdr-3-5, qKdr-3-6 and qKdr -6-1) were identified based on the mean values of GDR across 2 years, which could explain 14.29, 9.12, 6.03, 6.64, 7.28 and 13.06 % of phenotype variance, respectively. The additive effects of these QTLs were all positive with GDR being counted for 33.73 %, indicating that these alleles were all from parent Ji846, and could enhance GDR after physiological maturity by 0.0705, 0.0576, 0.0462, 0.0481, 0.0502 and 0.0667, respectively. In Shuangcheng, seven QTLs including qKdr-2-1, qKdr-3-1, qKdr-3-3, qKdr-3-6, qKdr-5-1, qKdr-8-1 and qKdr-8-2, were detected, which could explain 16.28, 7.05, 8.29, 9.49, 6.65, 7.07 and 5.89 % of phenotype variance, respectively. The additive effects of the total seven QTLs mentioned-above were all positive with GDR being counted for 60.72 %, which showed that these alleles were all from parent Ji846, and could enhance GDR after physiological maturity by 0.0866, 0.0562, 0.0621, 0.0657, 0.0543, 0.0562 and 0.0518, respectively.

SSR and AFLP linkage map and distribution of QTLs associated with GDR after physiological maturity
A total of 10 QTLs were detected with mean values of GDR at two-locations and two-years, which were qKdr-2-1, qKdr-2-2, qKdr-2-3, qKdr-3-1, qKdr-3-3, qKdr-3-4, qKdr-3-6, qKdr-6-2, qKdr-8-1 and qKdr-8-2, with 5.62, 4.04, 4.40, 4.26, 7.54, 4.54, 6.39, 5.40, 3.49 and 4.09 % of phenotypic variance, respectively. The additive effects of these QTLs were all positive with GDR being counted for 49.77 %, indicating that these alleles were mainly from parent Ji846. Of these QTLs, qKdr-2-1 and qKdr-3-6 located between phi090-umc1560 and phi046-bnlg1754, were stably detected in both Harbin and Shuangcheng, with phenotype variance of 15.59 and 10.28 %, and could enhance GDR by 0.0731, and 0.0585, respectively. QTL qKdr-3-4, with slight enhancement on phenotype variance, was consistent in Harbin, but it was not detected in Shuangcheng. QTL qKdr-3-3, qKdr-8-1 and qKdr-8-2 were consistent in Shuangcheng only with slight change on phenotype variance (+1.98, −1.09 and −0.24 %), but they were not detected in Harbin. It was difficult to use the four new QTLs detected by the mean values of GDR in the two locations because the four QTLs were only detected in one location. Probably they were easily influenced by environments or expressed unstably because of the regulation of other genes.
Discussion
The availability of a reliable methodology to measure GDR under field conditions is a bottleneck in selection for field grain drying rate (FDR) (Kang et al. 1978). Currently, there are lot of methods to measure GDR of maize. The main difference lies in the field sampling methods, including taking off a single ear, using the field electron detector of corn ear moisture (Kang and Zuber 1989) and taking the ear samples on different plants in the same pollination date (Zhang et al. 2007a). Single ear sampling method is to select some kernels from the ear’s middle part to measure GM in a number of days after the pollination, and the ear can be recovered to the original state after sampling to ensure their normal growth and development. The advantage of this method is that the dynamic data can be measured and sampling difference from different plants can be effectively avoided. However, the bract of the ear was destroyed and dehydration of maize grain is not processed under natural growth conditions using this method. The method of sampling different plants pollinated in the same time can fully ensure the sampling times for the ears, and the same developmental stages of the selected ears. GM can be measured in different development stages of the same materials, thereby the measured results is reliable. The disadvantages of this method are large number of samples, more technical steps, and overload work. By our results, this method is feasible.
Currently, many researches focused on the differences of GDR of different materials, heritability and gene action mode. The results showed that natural GDR after physiological maturity was quite different among the varieties, which was controlled by genes and showed mainly additive effects (Nass and Crane 1970; Jin et al. 1997). This information suggested that the GDR was mainly controlled by additive effects genes and was highly heritable, and breeding varieties with higher GDR was possible (Nass and Crane 1970).
In this study, different GDR of 280 RILs was evaluated over 2-locations and 2-years. The GDR values of most lines were between the two parents, showing the normal distribution. High significance between years or lines, interactions among the two element and the three elements plus locations were analyzed using the combined analysis of variance of the field GDR across the two locations in 2007 and 2008, respectively. The result indicated that the genetic basis of natural GDR after physiological maturity were really diverse in different families. Natural GDR after physiological maturity was highly heritable, as revealed by the estimates of heritability and their confidence intervals. The result of present study further validated that GDR was a quantitative trait with stable inheritance and can be effectively selected.
In this study, the average distance between two markers was about 16.1 cM, included nine markers in the fifth and seventh chromosome, respectively. Although there were no markers in some areas of the map, the basic framework of maize chromosomes had been constructed and can be used for QTL location and effect analysis. A large number of linkage map showed that markers in the genome was not uniformly distributed. Even in the map of small average distance, there was still a large gap area. However, Yano et al. (1997) found that there was no significant difference detecting the position and effect of QTL, when the distance between two markers was 5–21 cM, and increasing the number of markers has little significance in discovery of new QTL. Some studies suggested that the estimation of QTLs location and effects was relatively accurate so long as the distance between the markers was no more than 50 cM (Rebaï et al. 1995). The appropriate marker distance (such as 15 cM) was helpful in detecting QTL, and too large or too small distance was not suitable (He et al. 2001). So the map constructed in this study could be used for initial QTL identification, and it also proved that SSR and AFLP markers were suitable for genetic map constructing.
The 14 QTLs were detected on the 2, 3, 4, 5, 6, 8, and 10th chromosome of maize in this study. QTL qKdr-5-1, detected in Shuangcheng, was consistent to one QTL identified by Sala et al. (2006) using 181 F2:3 population, which located in linkage group 5, controlling GDR 1 week before harvest to harvest. The location of qKdr-8-1 and qKdr-8-2, identified in Shuangcheng across 2 years was close to the QTL on linkage group 8, and both had additive effect, which meant that the QTLs for GDR could be stably expressed. However, no QTL on linkage group 1, and new QTLs on chromosome 2, 3, and 6 were detected in this study.
Only two QTLs (qKdr-2-1, qKdr-3-6), detected on chromosome 2 and 3, were stable across environments, which could explain 15.59 and 10.28 % of phenotypic variance, respectively. They may be major QTLs. Meanwhile, location of qKdr-2-1 in linkage group, was similar to one QTL affecting plant height and ear height (Zhang et al. 2007b; Tang et al. 2005), and qKdr-3-6 as that of QTL for grain protein and starch (Dudley et al. 2004). Hence, qKdr-2-1 and qKdr-3-6 was not only valuable for MAS of high GDR after physiological maturity in maize, but also instructive in gene pooling in high yield and quality maize breeding.
Abbreviations
- QTL:
-
Quantitative trait locus
- GDR:
-
Grain drying rate
- CTAB:
-
Hexadecyl trimethyl ammonium bromide
- SSR:
-
Simple sequence repeats
- AFLP:
-
Amplified fragment length polymorphism
References
Basten CJ, Weir BS, Zeng ZB (1996) QTL cartographer: a manual and tutorial for QTL mapping. Department of Statistics, North Carolina State University, Raleigh
Cai RS (1995) Progress on maize grain moisture. Foreign Agric Crop 3:53–54
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138(3):963–971
Crane PL, Miles SR, Newman E (1959) Factors associated with varietal differences in rate of field differences in rate of field drying in corn. Agron J 51:318–320
Dijak M, Modarres AM, Hamilton RI, Dwyer LM, Stewart DW, Mather DE, Smith DL (1999) Leafy reduced-stature maize hybrids for short-season environments. Crop Sci 39:1106–1110
Doerge RW, Churchill GA (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142(1):285–294
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Dudley JW, Dijkhuizen A, Paul C, Coates ST, Rocheford TR (2004) Effects of random mating on marker-QTL associations in the cross of the Illinois high protein-Illinois low protein maize strains. Crop Sci 44:1419–1428
Hallauer AR, Russell WA (1960) Effects of weather factor on grain moister reduction from silking to physiologic maturity in corn. Agronomy 26:225–229
He XH, Xu CW, Kua JM, Gu SL, Li T (2001) Principal factor affecting the power of detection and accuracy of QTL mapping. Acta Agronomica Sinica 27:469–475
Jin Y, Wang ZH, Zhang YL, Wang SH, Wang YS (1997) Difference analysis on the natural dry rate of kernel after wax ripening in maize hybrids. J Northeast Agric Univ 28(1):29–32
Kang MS, Zuber MS (1989) Combining ability for grain moister, and maturity in maize with yellow and white endosperms. Crop Sci 29:689–692
Kang MS, Zuber MS, Horrocks RD (1978) An electronic probe for estimating ear moisture content of maize. Crop Sci 18:1083–1084
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Etoh T (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lv XL, Lan JH, Zhang BS (2006) Study on ear moisture loss in maize. J Northwest Sci Technol Univ Agric For 34(2):47–51
McCouch SR, Cjp YG, Yano M (1997) Report on QTL nomenclature. Rice Cenet Newsl 14:11–13
Nass HG, Crane PL (1970) Effect of endosperm mutants on drying rate in corn. Crop Sci 10:141–144
Purdy JD, Crane PL (1967) Inheritance of drying rate immature corn. Crop Sci 7:294–297
Rebaï A, Goffinet B, Mangin B (1995) Comparing powers of different methods for QTL detection. Biometrics 51:87–99
Sala RG, Andrade FH, Camadro EL, Cerono JC (2006) Quantitative trait loci for grain moisture at harvest and field grain drying rate in maize (Zea mays L.). Theor Appl Genet 112:462–471
Senior LM, Manfred H (1993) Mapping maize microsatellites and polymerase chain reaction confirmation of the targeted repeats using a CT primer. Genome 36(5):884–889
Sweeney PM, Martin SK, Clucas CP (1994) Indirect inbred selection to reduce grain moisture in maize hybrids. Crop Sci 34:391–396
Tang H, Yan JB, Huang YQ, Zheng YL, Li JS (2005) QTL analyses for five agronomic traits in maize. Acta Genet Sinica 32(2):203–209
Troyer AF, Ambrose WB (1971) Plant characteristics affecting field drying rate of ear corn. Crop Sci 11:529–531
Wang ZH, Zhang ZC, Chang H, Jin Y, Wang LF (2001) Analysis of physiological mature stage and kernel naturally dry down rate in 38 corn inbred lines in heilongjiang. J Maize Sci 9(2):53–55
Yang JP, Rong YZ, Huang LJ, Tang HT, Xiang DQ, Dai JR (2004) Construction of the frame molecular linkage map in maize. Acta Agronomica Sinica 30(1):81–87
Yano M, Harushima Y, Nagaruma Y, Kurata N, Minobe YT, Sasaki Y (1997) Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theor Appl Genet 95:1025–1032
Zhang LG, Wang ZH, Zhang L, Wang X, Liu XJ, Chen XC, Li B, Zhang Y (2007a) Analysis of combining ability of kernel dehydration rate after physiological maturity in maize. Crops 3:21–22
Zhang ZM, Zhao MJ, Rong YZ, Pan GT (2007b) SSR linkage map construction and qtl identification for plant height and ear height in maize (Zea mays L.). Acta Agron Sinica 33(2):341–344
Zhao SJ, Wu HM (2002) Analysis of kernel naturally dry-down rate of corn hybrids. J Jilin Agric Sci 27(5):24–26
Acknowledgments
Financial support for this work was provided in part by the Natural Science Foundation of China (30571166), Post-doctoral research start-up capital (Post Doc Research Found BH-Q06099) and the Program for the graduate training of Heilongjiang province.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Z., Wang, X., Zhang, L. et al. QTL underlying field grain drying rate after physiological maturity in maize (Zea Mays L.). Euphytica 185, 521–528 (2012). https://doi.org/10.1007/s10681-012-0676-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-012-0676-2