
Abstract
The morphological traits of leaves, such as size and shape, are major determinants of plant architecture and strongly affect high yield performance. To understand the molecular mechanism governing flag leaf length, we analyzed quantitative trait loci (QTLs) affecting flag leaf length by employing 176 F2 individuals derived from a cross between two japonica rice cultivars: Shennong265 (SN265) and Lijiangxintuanheigu (LTH). We identified qFLL3, qFLL6 and qFLL9 from this F2 population. Flag leaf length was increased by SN265 alleles at qFLL3 and qFLL6, but by LTH allele at qFLL9. In order to eliminate the influence of qFLL3 and qFLL6, one single residual heterozygous plant for qFLL9 region, RH-qFLL9, was selected based on the genotypes of 114 simple sequence repeat (SSR) markers and used as the parent of a segregating population. Using this segregating population of 889 plants, this region was narrowed down to an interval between RM24423 and RM24434. According to the rice annotation project database, there are 17 predicted genes in the 198-kb target region.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Grain yield of rice is largely determined by the top three leaves (Oryza sativa L.). The top three leaves on a stem are the primary source of carbohydrate production. Particularly, the flag leaf produces 40–60% of carbohydrates accumulated into grains, which finally determines grain yield (Gladun and Karpov 1993a; Foyer 1987; Kholupenco et al. 1996; Gladun and Karpov 1993b).
Li et al. (1998) used an F2:4 population and 115 well-distributed restriction fragment length polymorphism (RFLP) markers to analyze the genetic basis underlying the relationship between source leaves (the top two leaves) and sink capacity, and discovered that QLl3a, QLl3b, QLl7 and QLl9 controlling leaf length are located on chromosomes 3, 7 and 9, while QLw3, QLw4 and QLw8 controlling leaf width are located on chromosomes 3, 4 and 8. Kobayashi et al. (2003) analyzed QTLs affecting flag leaf development and mapped nine QTLs affecting flag leaf length and six QTLs controlling flag leaf width. Cui et al. (2003) mapped seven QTLs affecting leaf area. Rice constitutively wilted 1 (cow1) mutant exhibits the narrow and rolling leaf phenotype, and COW1 gene encodes a flavin-containing monooxygenase (FMO) which belongs to a new member of the YUCCA protein family (Woo et al. 2007). Fujino et al. (2008) identified a spontaneous mutation of narrow leaf, termed narrow leaf 7 (nal7), which displayed significantly decreased leaf blade width. The NAL7 gene also encodes a FMO and shows sequence similarity to the YUCCA gene family which catalyzed tryptamine to create N-hydroxytryptamine in auxin biosynthesis. Further analysis indicated that NAL7 is involved in auxin biosynthesis (Fujino et al. 2008). Qi et al. (2008) characterized a classic rice dwarf mutant named narrow leaf1 (nal1) which exhibits a characteristic phenotype of narrow leaves. nal1 encodes a plant-specific protein with unknown biochemical function, affects polar auxin transport as well as the vascular patterns of rice plants and plays an important role in control of lateral leaf growth. Compared with the greater available information regarding the regulation mechanism for leaf width, the molecular mechanism governing flag leaf length remains obscure.
In this paper, we identified three QTLs affecting flag leaf length and conducted fine mapping of one of the three detected QTLs, qFLL9, to a 198-kb interval on chromosome 9. These results provide more information for better understanding of the molecular mechanism governing flag leaf length.
Materials and methods
Plant materials
To identify QTLs more easily, we selected and crossed parental japonica varieties that showed highly significant differences in flag leaf length: Lijiangxintuanheigu (LTH) with a very long flag leaf (43.2 ± 3.5 cm) and Shennong265 (SN265) with a short flag leaf (22.2 ± 3.3 cm), to produce an F2 population of 176 plants. QTL analysis for flag leaf length was carried out based on this F2 population.
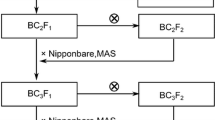
For fine mapping of qFLL9 as described later, five individuals from the F2 population were selected according to QTL analysis results. Foreground and background selection were conducted in the 125 F5 plants. Consequently, one F5 plant, which was heterozygous at the qFLL9 region on chromosome 9 and homozygous in other regions (termed RH-qFLL9), was selected based on genotyping results of 114 SSR markers. A segregating population of 899 individuals for the region of qFLL9 was developed. The scheme for constructing the RH-qFLL9 plant used in this study is summarized in Fig. 1.

Breeding scheme for the selection of the RH-qFLL9 plant in this study
Cultivation and trait measurements
The 176 F2 plants and 889 segregating plants from RH-qFLL9 were laid out in a field for phenotypic evaluation in the rice growing seasons of 2006 and 2009, respectively, on the experimental farm of the Rice Research Institute of Shenyang Agricultural University, Shenyang, China. The sowing date was April 12, and 15 plants per line with 2 LTH plants at each end to remove edge effects were transplanted on May 18, with 13.3 cm between plants and 30 cm between rows. Field management essentially followed standard agricultural practice. Fertilizers applied were 60, 90 and 90 kg/ha of N, P2O5 and K2O, respectively.
The parents and the two segregating populations showed a few days variation in heading date, therefore all plants were planted and investigated at the same time. Flag leaf length was defined as length from collar to top of leaf. At 25 days after heading, mean values for flag leaves on three main stems of each plant were used for data collection.
Genomic DNA extraction and PCR amplification
Total genomic DNA was extracted from young leaves of a single plant using the cetyltrimethylammonium bromide (CTAB) method (Murray and Thompson 1980). The SSR markers, developed by Temnykh et al. (2001) and Huang and Zhang (2003), were denoted by “RM” and “PM”, respectively. DNA amplification was performed using a GeneAmp PCR system 9700 thermocycler (Perkin Elmer Cetus, Norwalk, CT). Each reaction of 15 μl PCR mixture contained 20 ng genomic DNA, 50 mM KCl, 10 mM Tris–HCl (pH 8.8), 0.1% Triton-X, 1.5 mM MgCl2, 200 μM each of dNTPs, 0.2 μM of each primer, 5% (v/v) dimethyl sulphoxide and 0.5 U Taq DNA polymerase (Tiangen Biotech, Beijing, China). Amplification conditions consisted of initial denaturation at 94°C for 5 min, 35 cycles of 94°C for 45 s, 55–60°C for 45 s and 72°C for 1 min, followed by final extension at 72°C for 5 min. To detect polymorphisms of markers, polymerase chain reaction (PCR) products were separated on 6% polyacrylamide denaturing gels, and DNA fragments were detected by silver staining (Panaud et al.1996).
Genetic linkage map construction
The linkage map was constructed using the Mapmaker/Exp 3.0 program (Lander et al. 1987) on a personal computer. The command “GROUP” was used to verify the linkage group (LOD = 3, max distance = 50 cM), and the order of the linkage groups was determined by using the “ORDER” and “RIPPLE” commands. The “TRY” command was used to insert marker. The Kosambi function was used to calculate genetic distances.
QTL analysis and fine mapping of qFLL9
F2 population
Mapping of QTLs and estimation of their effects were carried out for flag leaf length by using the approach of composite interval mapping (CIM) in the computer package Windows QTL Cartographer version 2.5 (Wang et al. 2007). For CIM, a 2-cM window size was used for genome scans. The permutation method was used to obtain the thresholds of the experiment based on 1,000 runs of randomly shuffling the trait values (P = 0.05). The peak points of the LOD in the linkage map were taken as the putative positions of the QTLs, and additive effects of these QTLs were taken from the points showing the largest LOD. The relative contribution of a genetic component was calculated as the proportion of phenotypic variation explained by that component (Zeng 1994).
Segregating population from RH-qFLL9
For fine mapping of qFLL9, the bulked-extreme and recessive-class approach as described by Zhang et al. (1994) was used to calculate recombination frequencies between qFLL9 and molecular markers in the 187 homozygous plants with long flag leaf out of 889 segregating plants from RH-qFLL9. Thus, the recombination frequency = (N 1 + N 2/2)/N, where N is the total number of long-flag-leaf plants, N 1 is the number of long-flag-leaf plants with the marker genotype of the short-flag-leaf parent and N 2 is the number of long-flag-leaf plants with heterozygous marker genotype.
Results
Construction of linkage map and QTL mapping of flag leaf length using 176 F2 plants
An SSR linkage map of 114 marker loci was constructed (Fig. 2). All marker positions were in good agreement with previous maps (www.gramene.org). The mapped markers covered 12 rice chromosomes in 1397.5 cM of genetic distance with an average of 12.3 cM between two markers. The total genetic distance in the present population was 91.4% of that in the Japanese Rice Genome Research Program (RGP) map according to location information from www.gramene.org.

Graphical genotype of the RH-qFLL9 plant determined using 114 SSR markers. Solid and open bars represent SN265 segments and LTH segments, respectively. Putative location of each QTL is circled, and the size of each circle indicates the relative magnitude of their genetic effect
We mapped three QTLs for flag leaf length in this F2 population (Table 1). Among them, a major QTL for flag leaf length, qFLL9, was located between RM434 and RM242 on chromosome 9, at which the LTH allele contributed to long flag leaf (Fig. 3a). The additive effect of qFLL9 was –9.23. The amount of phenotypic variation explained by qFLL9 was 42.9% (Table 1).

Genetic map of qFLL9 controlling flag leaf length. The values between markers are mean recombination frequencies. a Location of qFLL9 on rice chromosome 9 estimated using 176 F2 plants. b High-resolution linkage map of the qFLL9 region produced based on 889 segregating plants from RH-qFLL9
Phenotypic performance of flag leaf length in the segregating population from RH-qFLL9
The RH-qFLL9 plant was homozygous at all SSR loci except for the region of qFLL9 on the long arm of chromosome 9 (Fig. 2). This enabled us to handle the qFLL9 locus as a single Mendelian factor in the segregating population. In fact, clear monogenic segregation for flag leaf length was observed in Fig. 4b, in apparent contrast to the results in Fig. 4a. We found that the heterozygous RH-qFLL9 plant had short flag leaf, and the segregation ratio of plants with short flag leaf and long flag leaf in the segregating population was 658:231 = 2.85:1.00, fitting well to the 3:1 ratio (χ 2 = 0.4593, P > 0.05). These results revealed that flag leaf length was controlled by a single gene and that long flag leaf is a recessive trait in this population.

Frequency distribution of flag leaf length in the F2 population from SN265/LTH (a) and a segregating population derived from RH-qFLL9 (b)
Fine mapping of qFLL9
For fine mapping of qFLL9, 187 homozygous plants with long flag leaf from the RH-qFLL9 plant and 18 new molecular markers surrounding qFLL9 were used to calculate the recombination frequency. Of these markers, three (RM24424, RM24428 and RM24432) were co-segregating completely with qFLL9 (Fig. 3b). Finally qFLL9 was mapped between RM24423 (16,965,639 bp) and RM24434 (17,163,735 bp) (http://rapdb.dna.affrc.go.jp/). qFLL9 was narrowed down to a 198-kb genomic region according to the information of these two markers. According to the rice annotation project database (http://rapdb.dna.affrc.go.jp/), there are 17 predicted genes in the 198-kb target region (Table 2). The functional information of these predicted genes was obtained from the UNIPROT protein knowledgebase (http://www.uniprot.org/).
Discussion
For fine mapping of QTL, it is necessary to carry out high-resolution linkage analysis using a large number of plants that segregate only around the QTL being investigated. Near-isogenic, introgression and chromosome segment substitution lines (Tian et al. 2006; Yu et al. 2007; Ebitani et al. 2005; Tan et al. 2008) developed by advanced backcrossing with marker-assisted selection have been widely used. Alternatively, a segregating population from a single plant with a heterozygous target QTL region on a homozygous background has increasingly frequently been used in recent years owing to its simple development (Yamanaka et al. 2005; Tuinstra et al. 1997). In this study, we used 889 individuals derived from the RH-qFLL9 plant to dissect the QTL affecting flag leaf length down to a single Mendelian factor. Because of the similar genetic background of the two parents (both being japonica rice varieties with small diversity), it is difficult to develop enough molecular markers to narrow down the target region. There are two approaches to resolve this problem; one is to increase the population size, and the other is to change the parent SN265 to an indica rice variety with short flag leaf. We are now taking these two approaches to obtain more recombination in the target region.
The most significant finding in our study was the delimitation of the qFLL9 gene to a DNA fragment of approximately 198-kb in length. Seventeen predicted genes were detected in the target region, including three genes (AK107584, AK111616 and J065094C22) related to cytochrome P450. Cytochrome P450 proteins are well known to be heme-binding enzymes, with monooxygenase activity such as oxidation, hydroxylation, isomerization and dehydration of various kinds of compounds (Nelson et al. 1996). D2/CYP90D2 and D11/CYP724B1 with shortening of clum, grain length and leaf length of rice have been already cloned encoding cytochrome P450 which play a role in brassinosteroid biosynthesis (Hong et al. 2003; Tanabe et al. 2005). Zhu et al. (2006) isolated an elongated uppermost internode mutation and revealed that the Eui gene causing this mutation encodes a previously uncharacterized P450, CYP714D1, as a gibberellins catabolism gene. Therefore, these three genes might be associated with control of flag leaf length. On the other hand, AK101247, whey acidic protein and core region domain containing protein, was identified as DENSE ELECT PANICLE 1 (DEP1) of rice, reducing the length of the inflorescence internodes, increasing the number of grains per panicle and grain yield and causing semi-dwarf stature. The parent material SN265 used in this study has the dep1 gene. Although dep1 is expressed in root, leaf, culm, inflorescence meristem and young inflorescence, it is expressed strongly in both the inflorescence meristem and the intercalary meristem but not in leaf (Huang et al. 2009). So, we considered that dep1 is different from qFLL9. Moreover, AK119560, similar to aldehyde dehydrogenase family seven member A1 and AK066748, similar to oryzain γ-chain precursor is expressed only in fruits of apple and seeds of soybean (Watanabe et al. 1991; Yamada et al. 1999). AK100287, similar to Axi 1 (auxin-independent growth promoter)-like protein, is expressed in chloroplast or Golgi apparatus (http://www.uniprot.org/). According to their organ specificity of gene expression, the latter four genes (AK101247, AK119560, AK066748 and AK100287) would not be target genes of qFLL9. Considering the organ specificity of gene expression and the molecular function information from the protein knowledgebase, AK107584, AK111616 and J065094C22 might be the most likely candidate genes, but we do not have enough data [polymorphisms or expression levels of these genes in leaves by reverse-transcription (RT)-PCR or quantitative real-time RT-PCR] to rule out the other ten candidate genes.
References
Cui KH, Peng SB, Xing YZ, Yu SB, Xu CG, Zhang Q (2003) Molecular dissection of the genetic relationships of source, sink and transport tissue with yield traits in rice. Theor Appl Genet 106:649–658
Ebitani T, Takeuchi Y, Nonoue Y, Yamamoto T, Takeuchi K, Yano M (2005) Construction and evaluation of chromosome segment substitution lines carrying overlapping chromosome segments of indica rice cultivar ‘Kasalath’ in a genetic background of japonica elite cultivar ‘Koshihikari’. Breed Sci 55:65–73
Foyer CH (1987) The basis for source-sink interaction in leaves. Plant Physiol Biochem 25:649–657
Fujino K, Matsuda Y, Ozawa K, Nishimura T, Koshiba T, Fraaije MW, Sekiguchi H (2008) NARROW LEAF 7 controls leaf shape mediated by auxin in rice. Mol Genet Genom 279:499–507
Gladun IV, Karpov EA (1993a) Distribution of assimilates from the flag leaf of rice during the reproductive period of development. Russ J Plant Physiol 40:215–219
Gladun IV, Karpov EA (1993b) Production and partitioning of assimilates between the panicle and vegetative organs of rice after flowering. Russ J Plant Physiol 40:728–773
Hong Z, Ueguchi-Tanaka M, Umemura K, Uozu S, Fujioka S, Takatsuto S, Yoshida S, Ashikari M, Kitano H, Matsuoka M (2003) A rice brassinosteroid-deficient mutant, ebisu dwarf (d2), is caused by a loss of function of a new member of cytochrome P450. Plant Cell 15:2900–2910
Huang CF, Zhang GQ (2003) Development of position-specific microsatellite markers and molecular mapping of insect resistant genes in rice (Oryza sativa L.). Mol Plant Breed 1:572–574
Huang X, Qian Q, Liu Z, Sun H, He S, Luo D, Xia G, Chu C, Li J, Fu X (2009) Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet 41:494–497
Kholupenco IP, Burundukova OL, Zhemchugova VP, Voronkova NM, Chernoded GK (1996) Source-sink relations in Far-Eastern rice cultivars as related to their productivity. Russ J Plant Physiol 43:141–148
Kobayashi S, Fukuta Y, Morita S, Sato T, Osaki M, Khush GS (2003) Quantitative trait loci affecting flag leaf development in rice (Oryza sativa L.). Breed Sci 53(3):255–262
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newberg L (1987) MAPMAKER: an interactive computer for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–182
Li Z, Pinson SRM, Stansel JW, Paterson AH (1998) Genetic dissection of the source-sink relationship affecting fecundity and yield in rice (Oryza sativa L.). Mol Breed 4:419–426
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8:4321–4325
Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC, Nebert DW (1996) P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6:1–42
Panaud O, Chen X, McCouch SD (1996) Development of micro-satellite marker and characterization of sample sequence lengthen polymorphism (SSLP) in rice (Oryza sativa L). Mol Gen Genet 252:597–607
Qi J, Qian Q, Bu Q, Li S, Chen Q, Sun J, Liang W, Zhou Y, Chu C, Li X, Ren F, Palme K, Zhao B, Chen J, Chen M, Li C (2008) Mutation of the rice Narrow leaf 1 gene, which encodes a novel protein, affects vein patterning and polar auxin transport. Plant Physiol 147:1947–1959
Tan L, Li X, Liu F, Sun X, Li C, Zhu Z, Fu Y, Cai H, Wang X, Xie D, Sun C (2008) Control of a key transition from prostrate to erect growth in rice domestication. Nat Genet 40:1360–1364
Tanabe S, Ashikari M, Fujioka S, Takatsuto S, Yoshida S, Yano M, Yoshimura A, Kitano H, Matsuoka M, Fujisawa Y, Kato H, Iwasaki Y (2005) A novel cytochrome P450 is implicated in brassinosteroid biosynthesis via the characterization of a rice dwarf mutant, dwarf11, with reduced seed length. Plant Cell 17:776–790
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–5142
Tian F, Li DJ, Fu Q, Zhu ZF, Fu YC, Wang XK, Sun CQ (2006) Construction of introgression lines carrying wild rice (Oryza rufipogon Griff.) segments in cultivated rice (Oryza sativa L.) background and characterization of introgressed segments associated with yield-related traits. Theor Appl Genet 112:570–580
Tuinstra MR, Ejeta G, Goldsbrough PB (1997) Heterogeneous inbred family (HIF) analysis: a method for developing nearisogenic lines that differ at quantitative trait loci. Theor Appl Genet 95:1005–1011
Wang S, Basten CJ, and Zeng ZB (2007) Windows QTL cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm
Watanabe H, Abe K, Emori Y, Hosoyama H, Arai S (1991) Molecular cloning and gibberellin-induced expression of multiple cysteine proteinases of rice seeds (oryzains). J Biol Chem 266:16897–16902
Woo YM, Park HJ, Su’udi M, Yang JI, Park JJ, Back K, Park YM, An G (2007) Constitutively wilted 1, a member of the rice YUCCA gene family, is required for maintaining water homeostasis and an appropriate root to shoot ratio. Plant Mol Biol 65:125–136
Yamada K, Mori H, Yamaki S (1999) Identification and cDNA cloning of a protein abundantly expressed during apple fruit development. Plant Cell Physiol 40:198–204
Yamanaka N, Watanabe S, Toda K, Hayashi M, Fuchigami H, Takahashi R, Harada K (2005) Fine mapping of the FT1 locus for soybean flowering time using a residual heterozygous line derived from a recombinant inbred line. Theor Appl Genet 110:634–639
Yu B, Lin Z, Li H, Li X, Li J, Wang Y, Zhang X, Zhu Z, Zhai W, Wang X, Xie D, Sun C (2007) TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J 52:891–898
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang Q, Shen BZ, Dai XK, Mei MH, Saghai Maroof MA, Li ZB (1994) Using bulked extremes and recessive class to map genes for photoperiod-sensitive genic male sterility in rice. Proc Natl Acad Sci U S A 91:8675–8679
Zhu Y, Nomura T, Xu Y, Zhang Y, Peng Y, Mao B, Hanada A, Zhou H, Wang R, Li P, Zhu X, Mander LN, Kamiya Y, Yamaguchi S, He Z (2006) ELONGATED UPPERMOST INTERNODE encodes a cytochrome P450 monooxygenase that epoxidizes gibberellins in a novel deactivation reaction in rice. Plant Cell 18:442–456
Acknowledgments
This research was supported by the National Natural Science Foundation of China (30871468) and the National 973 Program of China (2009CB126007).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jiang, S., Zhang, X., Wang, J. et al. Fine mapping of the quantitative trait locus qFLL9 controlling flag leaf length in rice. Euphytica 176, 341–347 (2010). https://doi.org/10.1007/s10681-010-0209-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-010-0209-9