
Abstract
Primitive cottons (Gossypium spp.) represent resources for genetic improvement. Most primitive accessions are photoperiod sensitive; they do not flower under the long days of the U.S. cotton belt. Molecular markers were used to locate quantitative trait loci (QTLs) for node of first fruiting branch (NFB), a trait closely related to flowering time in cotton. An F2 population consisted of 251 plants from the cross of a day neutral cultivar Deltapine 61, and a photoperiod sensitive accession Texas 701, were used in this study. Segregation in the population revealed the complex characteristics of NFB. Interval mapping and multiple QTL mapping were used to determine QTLs contributing to NFB. Three significant QTLs were mapped to chromosome 16, 21, and 25; two suggestive QTLs were mapped to chromosome 15 and 16. Four markers associated with these QTLs accounted for 33% of the variation in NFB by single and multiple-marker regression analyses. Two pairs of epistasis interaction between markers were detected. Our results suggested that at least three chromosomes contain factors associated with flowering time for this population with epistasis interactions between chromosomes. This research represent the first flowering time QTL mapping in cotton. Makers associated with flowering time may have the potential to facilitate day neutral conversion of accessions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A narrow genetic base for domesticated Upland cotton (Gossypium hirsutum L.) exists in the major cotton producing countries such as U.S. (Van Esbroeck et al. 1998, Bowman, 2000), China (Liu et al. 2003), Australia (Multani and Lyon 1995), India (Rana and Bhat 2005), and Pakistan (Rahman et al. 2002). The narrow genetic base of U.S. domesticated cotton is due to a high degree of similarity among ancestral cultivars and limited sources of wild germplasm (Van Esbroeck et al. 1999) and may increase the crop’s vulnerability to pests, diseases, and environmental stresses (Holley and Goodman 1989).
The narrow genetic base has been considered as one of the major obstacles in cotton improvement (Meredith 2000). The utilization of primitive G. hirsutum germplasm is an important approach to acquire new alleles and broaden cotton’s genetic base. Perennial, primitive accessions of cotton have been described as an important source of useful genetic variability for cotton genetic improvement and may limit its vulnerability to pests, diseases, and environmental stresses (Percival 1987; Meredith 1991; McCarty and Jenkins 1993, 2004; McCarty et al. 1995, 2004a, b). Primitive G. hirsutum accessions have been reported to carry valuable alleles for resistance to cotton pests (Jenkins 1986) and pathogens (Haley and Wilhelm 1975; Percival 1987), variability for seed-oil content (Kohel 1978), and high fiber quality (McCarty et al. 2006). However, most primitive accessions are photoperiod sensitive; they do not flower under the long days of the U.S. cotton belt. Thus the photoperiod sensitivity is a major obstacle in the use of primitive germplasm in cotton breeding programs.
A phenotypic backcross breeding program has been undertaking by USDA-ARS to incorporate day-neutral genes into the primitive accessions for their utilization in breeding (McCarty et al. 1979). However, some day neutral converted accessions showed lower genetic diversity than their wild photoperiodic counterparts by molecular markers assay (Liu et al. 2000; Zhong et al. 2002). Linkage drags during backcrossing in the day neutral conversion program hampered the full recovery of alleles from the photoperiodic parent.
Few systematic genetic studies have been conducted for flower initiation in cotton. Although conventional genetic analysis concluded the inheritance of flowering response in G. hirsutum followed a multigenic pattern, the inheritance mode and the number of genes involved depended on the materials used (Lewis and Richmond 1957, 1960; Waddle et al. 1961; Kohel et al. 1974). Given the complex character of flower initiation in plants studied in detail (Koester et al. 1993; Lin et al. 1995; Yano et al. 1997; Börner et al. 2002; Komeda 2004), it is reasonable to hypothesize that multiple QTLs may jointly determine cotton flower initiation. Since the knowledge of the flowering mechanisms of cotton at the molecular level is limited and no genes/QTLs for flowering in Upland cotton have been localized on specific chromosomes in previous studies, such a study should provide insight to this vital function.
Node of first fruiting branch (NFB) is an indication of flowering time and a measure of relative photoperiodism. In cotton, it reflects the flowering character of stocks. Hutchinson (1959) concluded that fruiting branches are not formed until a certain number of nodes develop on the main stem. Ray and Richmond (1966) studied various morphological measures of earliness in cotton and considered NFB as the most reliable and practical measurement. Moreover, Low et al. (1969) suggested using the main-stem NFB as a criterion to measure earliness in consideration of its good heritability and correlation with final picking.
The objective of this study was to determine cotton flowering time QTLs reflected by node of first fruiting branch (NFB) in a primitive accession Texas 701(T701). This research provides the first investigation using QTL mapping for flowering time (NFB) in cotton.
Materials and methods
Plant materials and DNA isolation
Upland cotton cultivar ‘Deltapine 61’ (DPL61) (PI No. 607174) was crossed as female parent with short-day accession ‘Texas 701’ (T701) (PI No. 165329), at a cotton winter nursery in Tecoman, Mexico. T701 is a photoperiod sensitive accession, which does not produce flowers in Mississippi under the normal photoperiod of the cotton growing season, but flowers in Central Mexico. DPL61 is a day-neutral cultivar released by Delta and Pine Land Company. The F1 generation was self-pollinated at the winter nursery in Tecoman, Mexico. The F2 generation was planted in the second week of May at Mississippi State, MS (33.4 N, 88.8 W) in year 2000 following standard agronomic practices. The population contains 251 individuals and segregated for flowering response. The segregation of leaf (L 2 S) shape (abbreviated as LS), petal spot (R 2) (abbreviated as PS), and petal color (Y 1) (abbreviated as PC) of the F2 population were scored on Aug 21st, 2000 as Mendelian traits. Node of first fruiting branch (NFB) was classified for each plant on October 17th, 2000. NFB is the first node above the cotyledon node on the mainstem that a fruiting branch develops and produces fruiting buds that develop into flowers and fruit. Mauney (1986) reviewed the development of cotton fruiting structures. The first fruiting branch (sympodial) usually develops at mainstem node 6 to 7 and a fruiting branch will continue to arise at each vertical mainstem node for the remainder of the growing season for commercial Upland cotton. Genomic DNA was isolated from frozen young leaf samples using DNeasy Mini Kit (QIAGEN, Chatsworth, CA, USA) following the manufacture’s protocol.
Markers assay
A total of 1165 SSR primer pairs were used for parental lines polymorphism screening. These SSR primers included BNL (Research Genetics Co., Huntsville, AL, USA), JESPR (Reddy et al. 2001), MGHES (Qureshi et al. 2004), TMB (provided by USDA-ARS, Crops Germplasm Research Unit, College Station, TX), CIR (Nguyen et al. 2004), and NAU (Han et al. 2006). The marker acronym followed Nguyen et al. (2004). Capillary electrophoresis analysis was conducted with an ABI 3100 genetic analyzer (Applied Biosystems, Foster City, CA, USA) using fluorescently labeled SSR markers according to Gutierrez et al. (2002).
Selective determination of marker-trait association
The purpose of selective determination was to identify NFB tightly related markers among all the polymorphic ones. Two sub-groups each consisting of 15 (6%) F2 plants were selected to represent the two extreme groups, photoperiod sensitive and day neutral. All the 222 polymorphic primer pairs were screened against individuals within both groups. Pearson Correlation coefficients between genotyping and day neutrality trait were calculated by SAS 9.0 (SAS institute Inc., 1999) with the command ‘PROC CORR’. Markers with a P value of less than 0.05 were first chosen to screen the whole F2 population, and then the markers with P value of less than 0.1, less than 0.2, were used orderly to screen the full F2 population. After the construction of a tentative linkage map and the estimation of putative QTL positions and effects, more markers on the specific putative QTLs located chromosomes were added for genotyping.
Linkage and QTL analysis
Genetic linkage map was constructed by JOINMAP3.0 (Van Ooijen and Voorrips 2001). A minimum LOD score (log10 of the likelihood odds ratio) of 5 was set as a threshold to allocate marker locus into linkage groups, and a maximum recombination fraction of 0.40 was employed as general linkage criteria to establish linkage groups. The Kosambi function was used to order markers and to estimate map unit distances (Kosambi 1944). Segregation distortion at each marker locus was tested against the expected segregation ratios (1:2:1 for co-dominant markers and 3:1 for dominant markers) using a chi-square goodness of fit test. Linkage groups chromosomal assignment were achieved by CMD (Cotton Microsatellite Database, http://www.cottonmarker.org) inquiry, comparison to the published integrated molecular maps (Nguyen et al. 2004; Lacape et al. 2005; Han et al. 2006; Wang et al. 2006b; Guo et al. 2007), and our unpublished data.
Since the NFB distribution in the segregating population deviated from normality, several methodologies were used for trait and marker association analyses. Interval mapping, multiple QTL mapping (MQM), and non-parametric genome scan based on the Kruskal–Wallis (KW) analysis were performed with MAPQTL5.0 (Van Ooijen 2004). For interval mapping, genome wide LOD significance threshold (3.8) were calculated by 1,000 × permutation test (Van Ooijen, 1999), which restricted the occurrence of Type I statistical errors (false positives) to less than 5%. A suggestive QTL had LR value above 9.2 (equal to LOD score of 2.0) according to Lander and Kruglyak (1995). Markers with the highest LOD values were then used in various combinations as co-factors in multiple QTL models in MQM mapping. QTL graphs were made by MAPCHART 2.2 (Voorrips 2002). We followed the typical QTL nomenclature (McCouch et al. 1997) which is a designation of ‘q’ followed by an abbreviation of a trait name (NFB), then the chromosome on which it was located, and the number of the detected QTLs affecting the trait on the chromosome. All the markers on chromosome 15, 16, 21 and 25 were further tested for their association with NFB and epistasis using regression analyses by ‘PROC GLM’ command of SAS 9.0 (SAS Institute Inc., 1999).
Results
Performance of NFB
NFB for DPL61 ranged from 6 to 8 while T701 did not set any fruiting branches by harvest. The F1 plants had NFB ranging from 13 to 16 (setting flowers at main stem node 13–16) indicating NFB was controlled by partially dominant genes. In the F2 population, NFB ranged from 7 to 24 with a mean of 21 and standard deviation of 6.6. There were 15 (6%) plants with NFB from 7 to 9, 25 (10%) plants from 10 to 12, 63 (25%) from 13 to 24, and 148 (59%) plants that did not set any flowers by harvest. The distribution of NFB in the F2 population was skewed toward the photoperiod sensitive parent (T701). The skewness was −0.64, and the kurtosis was −1.29. Transgressive segregation was not observed. Plants with NFB ≤ 12 or 13 are generally considered as day neutral (the flower will set bolls which could be harvested by frost). Either NFB ≤ 12 or NFB ≤ 13, the segregation ratio carried by χ2 test indicated that at least two genes with epistasis controlling the NFB.
Selective determination of marker-trait association
Selective 10–20% of the population for genotyping is an effective and efficient way for QTL detection (Darvasi and Soller 1992; Barrett 2002). This procedure was employed to seek the most NFB related markers, increase power of QTL detection, and reduce cost. The phenotypic extreme plants used for selective determination consisted of about 12% of the population. Markers detected at significant level P < 0.01, 0.01 < P < 0.05, 0.05 < P < 0.1, and 0.1 < P < 0.2 were 7, 19, 21, and 32, respectively. All 47 markers significant at P < 0.1 level detected by selective determination were significant at P < 0.05 level in the full set population with nonparametric KW test. In addition, nine markers detected by selective determination by 0.1 < P < 0.2 significant level were also significant at P < 0.05 level by using the full population with nonparametric KW test (data not shown). This step ensured most of the NFB related markers were used for constructing the linkage map.
Linkage map construction
Ninety four SSR markers and three morphological markers were used to construct the genetic linkage map. Among them, 15 markers had distorted segregation (9 at α = 0.05 level, 1 at 0.01 level, 5 at 0.001 level). Seventy SSR makers and three morphological makers coalesced in 17 linkage groups covering 650.8 cM (centimorgans), approximately 14.5% of the cotton genome (Rong et al. 2004). The 17 linkage groups were assigned to 14 chromosomes/linkage groups. The marker order of our linkage map agreed with previous published maps (Jiang et al. 2000; Nguyen et al. 2004; Lacape et al. 2005; Han et al. 2006; Guo et al. 2007).
QTL analysis for NFB
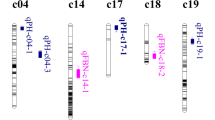
The location of significant QTLs and their flanking markers, LOD-1 and LOD-2 confidence intervals are presented in Table 1 and Fig. 1. By interval mapping, three significant QTLs for NFB were found on chromosome 16, 21, and 25, explaining 9.1, 19.3, and 10.2% of the phenotypic variation, with additive effect of −2.73, −3.39, and −2.37, respectively. Another two suggestive QTLs with small effects were detected on chromosome 15 and 16. Their LOD values were between 2.5 and 3.8, explained 8.1 and 4.9% of the phenotypic variation, with additive effects of 2.28 and −1.89, respectively. No QTLs were found within the A subgenome (chromosomes 1–13). MQM mapping results confirmed the three QTLs detected by interval mapping, except the interval for each QTL became much narrower (Table 1 and Fig 1.). The location of qNFB-c21-1 remained the same as interval mapping, but explained 17.3% of the phenotypic variation. The location of qNFB-c16-1 and qNFB-c25-1 estimated by MQM mapping shifted 2 and 6 cM from that in interval mapping. The phenotypic variation explained change to 7.8 and 15.2%, respectively. Results from both interval mapping and MQM mapping indicated that most alleles originated from T701 appeared to increase NFB. In addition to QTL mapping, non-parametric KW analysis was used for detecting markers associated with NFB. Results showed that chromosome 15, 16, 21, and 25 contained far more NFB associated markers than the other chromosomes (Fig. 1).

QTLs detected on chromosome15, 16, 21 and 25 for node of first fruiting branch (NFB) by interval mapping and Multiple QTL Mapping. LS2 represents super okra leaf shape (L 2 S); Stars indicate significant markers detected from KW non-parametric test at different significant level with *, **, and *** representing significant at P = 0.05, P = 0.01, and P = 0.001, respectively
Regression analysis
Of the markers used for single marker analysis, eight (Table 2) showed significant additive or dominant effects, and individually explained 4–11% of the phenotypic variation for NFB. Multiple marker regression analysis showed that four markers near the significant QTLs peak collectively explain 33% of the phenotypic variance for NFB (Table 2). They were JESPR102-110, JESPR251-72, BNL2812-146, and TMB0436-157. These markers could be tentatively used as the major indicator of flowering time in marker assistant selection (MAS). If we include the nearest markers to the two suggestive QTLs for multiple marker regression analysis, the total explained phenotypic variance would increase to 37%. Thus the two suggestive QTLs on chromosome 15 and 16 only contributed 4% of the phenotypic variance. Regression analysis also detected the epistasis interaction between two pairs of markers. One is JESPR102-110 (chromosome 16) and TMB0436-157 (chromosome 25); the other is JESPR102-110 (chromosome 16) and BNL2812-146 (chromosome 21). This suggested epistasis interactions might be present between qNFB-c16-1 and qNFB-c25-1 or between qNFB-c16-1 and qNFB-c21-1.
Discussion
Genes controlling flowering time in cotton
Limited research had been conducted on cotton flower initiation. In G. barbadense L., the day neutral trait was controlled by one gene pair and the short-day non-flowering response gene was dominant to the flowering gene (Lewis and Richmond 1960). In G. hirsutum, flower initiation was controlled by a complex of genes (Lewis and Richmond 1957, 1960; Waddle et al. 1961), controlled by partially dominant (Lewis and Richmond 1957; Waddle et al. 1961), or recessive genes (Lewis and Richmond 1960). Segregation patterns varied within different accessions. The partially dominant genetic mechanism detected in our F2 population agreed with the conclusion from Lewis and Richmond (1960). Chi-square test suggested day neutrality may be primarily controlled by two genes, but QTL mapping results indicated at least three significant QTLs and two suggestive QTLs involved in main stem fruiting branch initiation. The differences may be due to some QTLs started taking effect on NFB initiation after node 12/13. This might indicate that flowering time is a process not simply controlled by several genes, but by multiple genes that may start their function at different cotton development stages. Also, these genes probably form a complex network to interact with each other, which is supported by the potential epistasis QTLs found in this study.
QTL scattering pattern may be responsible for linkage drag
Maintaining the highest recovery rate of accession parent allele is crucial to broaden the genetic base of Upland cotton in a day neutral conversion program. Zhong et al. (2002) used AFLP markers to study five populations (F6, BC1F6, BC2F6, BC3F6, BC4F6) of day neutral plants derived from crossing and backcrossing of four accessions. The results showed the recovery of markers from the exotic parent did not increase significantly during additional backcross generations. Many AFLP markers tended to stay together as linked blocks when selecting the day neutral phenotype, which leads to linkage drag. Liu et al. (2000) assessed ninety-six day neutral converted accessions by SSR markers. The graphic genotype indicated poor recovery for some accessions. All these indicated linkage drag might locate near genes controlling day neutrality in the donor parents.
Our significant QTLs for NFB are on chromosomes 16, 21, and 25. These chromosomes also host QTLs for fiber quality related traits. Compared with previously published papers for QTLs responsible for cotton fiber quality (Paterson et al. 2003; Mei et al. 2004; He et al. 2005, 2007; Lacape et al. 2005; Shen et al. 2005, 2006, 2007; Wang et al. 2006a), most of the QTLs for NFB (photoperiod sensitivity) were linked with some QTLs related with fiber quality. Lacape et al. (2005) proposed that in cotton, there might be QTL-rich regions along a chromosome congruent between mapping populations, generations and locations. Wang et al. (2006a) suggested that QTLs influencing the quality of cotton fiber might exist in clusters. If genes responsible for NFB and genes related to fiber quality are linked, the selection of day neutral plants during conversion of accessions to day neutral lines would also select fiber and yield genes from the day neutral parents, instead of from the photoperiod sensitive accessions. This may be the reason for linkage drag observed by Zhong et al. (2002) and the relatively stable character of fiber quality traits across backcrossing generations (McCarty et al. 1998). Simultaneous selection of day neutral plants with desired fiber quality by molecular markers assistance may facilitate achieving the aim to maintain the maximum genetic diversity in a day neutral conversion program. This study firstly provided the information on the inheritance of flowering time in cotton by QTL mapping. It may have the potential to accelerate the simultaneous selection of day neutral plants with desired fiber quality and yield.
Abbreviations
- DPL61:
-
Deltapine 61
- LOD:
-
Logarithm of odds
- MQM:
-
Multiple QTL mapping
- NFB:
-
Node of first fruiting branch
- T701:
-
Texas accession 701
References
Barrett J (2002) Association studies. In: Camp NJ, Cox A (eds) Quantitative trait loci-methods and protocols. Humana Press, Totowa, NJ, pp 3–12
Bowman DT (2000) Attributes of public and private cotton breeding programs. J Cotton Sci 4:130–136
Börner A, Buck-sorlin GG, Hayes PM, Malyshev S, Korzun V (2002) Molecular mapping of major genes and quantitative trait loci determining flowering time in response to photoperiod in barley. Plant Breed 121:129–132
Darvasi A, Soller M (1992) Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor Appl Genet 85:353–359
Guo W, Cai C, Wang C, Han Z, Song X, Wang K, Niu X, Wang C, Lu K, Shi B, Zhang T (2007) A microsatellite-based, gene rich linkage map reveals genome structure, function, and evolution in Gossypium. Genetics 176:527–541
Gutierrez OA, Basu S, Saha S, Jenkins JN, Shoemaker DB, Cheatham CL, McCarty JC (2002) Genetic distance among selected cotton genotypes and its relationship with F2 performance. Crop Sci 42:1841–1847
Haley AB, Wilhelm S (1975) Comparative verticillium wilt reaction of wild races of Gossypium hirsutum and hybrids with upland cultivars. In: Brown JM (ed) Proceedings of the beltwide cotton producers conference, New Orleans, LA. 6–8 Jan 1975
Han Z, Wang C, Song X, Guo W, Guo J, Li C, Chen X, Zhang T (2006) Characteristics, development and mapping of Gossypium hirsutum derived EST-SSRs in allotetraploid cotton. Theor Appl Genet 112:430–439
He DH, Lin ZX, Zhang XL, Nie YC, Guo XP, Feng CD, Stewart JM (2005) Mapping QTLs of traits contributing to yield and analysis of genetic effects in tetraploid cotton. Euphytica 144:141–149
He DH, Lin ZX, Zhang XL, Nie YC, Guo XP, Zhang YX, Li W (2007) QTL mapping for economic traits based on a dense genetic map of cotton with PCR-based markers using the interspecific cross of Gossypium hirsutum × Gossypium barbadense. Euphytica 153:181–197
Holley RN, Goodman MM (1989) New sources of resistance to southern corn leaf blight from tropical hybrid maize derivatives. Plant Dis 73:562–564
Hutchinson JB (1959) The application of genetics to cotton improvement. Cambridge University Press, Cambridge
Jenkins JN (1986) Host plant resistance: Advances in cotton. In: Brown JM (ed) Proceedings of the beltwide cotton producers conference, Las Vegas, NV. 4–9 Jan 1986
Jiang C, Wright R, Woo S, Delmonte T, Paterson AH (2000) QTL analysis of leaf morphology in tetraploid Gossypium (cotton). Theor Appl Genet 100:409–418
Koester RP, Sisco PH, Stuber CW (1993) Identification of quantitative trail loci controlling days to flowering and plant height in two near isogenic loci of maize. Crop Sci 33:1209–1216
Kohel RJ (1978) Survey of Gossypium hirsutum L. germplasm collection of seed-oil percentage and seed characteristics. USDA Report ARS-S-187
Kohel RJ, Richmond TR, Lewis CF (1974) Genetics of flowering response in cotton. VI. Flowering behavior of Gossypium hirsutum L. and G. barbadense L. hybrids. Crop Sci 14:696–699
Komeda Y (2004) Genetic regulation of time to flower in Arabidopsis thaliana. Annu Rev Plant Biol 55:521–535
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lacape JM, Nguyen TB, Courtois B, Belot JL, Giband M, Gourlot JP, Gawryziak G, Roques S, Hau B (2005) QTL analysis of cotton fiber quality using multiple Gossypium hirsutum × Gossypium barbadense backcross generations. Crop Sci 45:123–140
Lander ES, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature Genet 11:241–247
Lewis CF, Richmond TR (1957) The genetics of flowering response in cotton. I. Fruiting behavior of Gossypium hirsutum var. marie-galante in a cross with a variety of cultivated American Upland cotton. Genetics 42:499–509
Lewis CF, Richmond TR (1960) The genetics of flowering response in cotton. II. Inheritance of flowering response in a Gossypium barbadense cross. Genetics 45:79–85
Lin YR, Schertz KF, Paterson AH (1995) Comparative analysis of QTLs affecting plant height and maturity across the Poaceae, in reference to an interspecific sorghum population. Genetics 141:391–411
Liu S, Cantrell RG, McCarty JC, Stewart JM (2000) Simple sequence repeat-based assessment of genetic diversity in cotton race stock accessions. Crop Sci 40:1459–1469
Liu WX, Kong FL, Guo ZL, Zhang QY, Peng HR, Fu XQ, Yang FX (2003) An analysis about genetic basis of cotton cultivars in China since 1949 with molecular markers. Acta Genet Sin 30:560–570 (in Chinese, English abstract)
Low A, Hesketh J, Muramoto H (1969) Some environmental effects on the varietal node number of the first fruiting branch. Cotton Growing Rev 46:181–188
Mauney JR (1986) Vegetative growth and development of fruiting sites. In: Mauney JR, Stewart JM (eds) Cotton physiology. The Cotton Foundation, Memphis, TN, pp 11–28
McCarty JC, Jenkins JN (1993) Registration of 79 day-neutral primitive cotton germplasm lines. Crop Sci 33:351
McCarty JC, Jenkins JN (2004) Primitive cotton germplasm: yield and fiber traits for 21 day-neutral acessions. Mississippi Agric For Exp Stn Res Rep 23(14):1–6
McCarty JC, Jenkins JN, Parrott WL, Creech RG (1979) The conversion of photoperiodic primitive race stocks of cotton to day-neutral stocks. Mississippi Agric For Exp Stn Res Rep 4(19):1–4
McCarty JC, Jenkins JN, Tang B (1995) Primitive cotton germplasm: variability for yield and fiber traits. Miss Agric and For Exp Stn Tech Bull 202:8 pp
McCarty JC, Jenkins JN, Wu J (2004a) Primitive accession derived germplasm by cultivar crosses as sources for cotton improvement: I. phenotypic values and variance components. Crop Sci 44:1226–1230
McCarty JC, Jenkins JN, Wu J (2004b) Primitive accession derived germplasm by cultivar crosses as sources for cotton improvement: II. genetic effects and genotypic values. Crop Sci 44:1231–1235
McCarty JC, Jenkins JN, Zhu J (1998) Introgression of day-neural genes in primitive cotton accessions: I. Genetic variances and correlations. Crop Sci 38:1425–1428
McCarty JC, Wu J, Jenkins JN (2006) Genetic diversity for agronomic and fiber traits in day-neutral accessions derived from primitive cotton germplasm. Euphytica 148:283–293
McCouch SR, Cho YG, Yano PE, Blinstrub M, Morishima H, Kinoshita T (1997) Report on QTL nomenclature. Rice Genet Newslett 14:11–13
Mei M, Syed NH, Gao W, Thaxton PM, Smith CW, Stelly DM, Chen ZJ (2004) Genetic mapping and QTL analysis of fiber-related traits in cotton (Gossypium). Theor Appl Genet 108:280–291
Meredith WR (1991) Contributions of introductions to cotton improvement. In: Shands HL, Weisner LE (eds) Use of plant introductions in cultivar development. Part I. Crop Science Society of America, Madison, WI, p 127–146
Meredith WR (2000) Cotton yield progress—why has it reached a plateau. Better Crops 84:6–9
Multani DS, Lyon BR (1995) Genetic fingerprinting of Australian cotton cultivars with RAPD markers. Genome 38:1005–1008
Nguyen TB, Giband M, Brottier P, Risterucci AM, Lacape JM (2004) Wide coverage of the tetraploid cotton genome using newly developed microsatellite markers. Theor Appl Genet 109:167–175
Paterson AH, Saranga Y, Menz M, Jiang CX (2003) QTL analysis of genotype × environment interactions affecting cotton fiber quality. Theor Appl Genet 106:384–396
Percival AE (1987) The national collection of Gossypium germplasm. Southern Cooperative Serials Bulletin 321:362 pp
Qureshi SN, Saha S, Kantety RV, Jenkins JN (2004) EST-SSR: a new class of genetic markers in cotton. J Cotton Sci 8:112–123
Rahman M, Hussain D, Zafar Y (2002) Estimation of genetic divergence among elite cotton cultivars-genotypes by DNA fingerprinting technology. Crop Sci 42:2137–2144
Rana MK, Bhat KV (2005) RAPD markers for genetic diversity study among Indian cotton cultivars. Curr Sci 88:1956–1961
Ray LL, Richmond TR (1966) Morphological measures of earliness of crop maturity in cotton. Crop Sci 6:527–531
Reddy OUK, Pepper AE, Ibrokhim A, Saha S, Jenkins JN, Brooks T, Bolek Y, El-Zik KM (2001) New dinucleotide and trinucleotide microsatellite marker resources for cotton genome research. J Cotton Sci 5:103–113
Rong J, Abbey C, Bowers JE, Brubaker CL, Chang C, Chee PW, Delmonte TA, Ding X, Garza JJ, Marler BS, Park C, Pierce GJ, Rainey KM, Rastogi VK, Schulze SR, Trolinder NL, Wendel JF, Wilkins TA, Williams-Coplin TD, Wing RA, Wright RJ, Zhao X, Zhu L, Paterson AH (2004) A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166:389–417
SAS Institute Inc. (1999) SAS software version 9.0. SAS Institute Inc., Cary, NC
Shen X, Guo W, Lu Q, Zhu X, Yuan Y, Zhang T (2007) Genetic mapping of quantitative trait loci for fiber quality and yield trait by RIL approach in Upland cotton. Euphytica 155:371–380
Shen X, Guo W, Zhu X, Yuan Y, Yu JZ, Kohel RJ, Zhang T (2005) Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol Breed 15:169–181
Shen X, Zhang T, Guo W, Zhu X, Zhang X (2006) Mapping fiber and yield QTLs with main, epistatic, and QTL × enviroment interaction effects in recombinant inbred lines of upland cotton. Crop Sci 46:61–66
Van Esbroeck GA, Bowman DT, Calhoun DS, May OL (1998) Changes in the genetic diversity of cotton in the USA from 1970 to 1995. Crop Sci 38:33–37
Van Esbroeck GA, Bowman DT, May OL, Calhoun DS (1999) Genetic similarity indices for ancestral cotton cultivars and their impact on genetic diversity estimates of modern cultivars. Crop Sci 39:323–328
Van Ooijen J, Voorrips RE (2001) JionMap® 3.0, Software for the calculation of genetic linkage maps. Plant Research International, Wageningen, the Netherlands
Van Ooijen JW (1999) LOD significance thresholds for QTL analysis in experimental populations of diploid species. Heredity 83:613–624
Van Ooijen JW (2004) MapQTL® 5, Software for the mapping of quantitative trait loci in experimental populations. Kyazma B.V., Wageningen, the Netherlands
Voorrips RE (2002) MapChart: Software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Waddle BM, Lewis CF, Richmond TR (1961) The genetics of flowering response in cotton. III. Fruiting behavior of Gossypium hirsutum race latifolium in a cross with a variety of cultivated American Upland cotton. Genetics 46:427–437
Wang B, Guo W, Zhu X, Wu Y, Huang N, Zhang T (2006a) QTL mapping of fiber quality in an elite hybrid derived-RIL population of upland cotton. Euphytica 152:367–378
Wang K, Song X, Han Z, Guo W, Yu J, Sun J, Pan J, Kohel R, Zhang T (2006b) Complete assignment of the chromosomes of Gossypium hirsutum L. by translocation and fluorescence in situ hybridization mapping. Theor Appl Genet 113:73–80
Yano M, Harushima Y, Nagamura Y, Kurata N, Minobe Y, Sasaki T (1997) Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theor Appl Genet 99:1025–1032
Zhong M, McCarty JC, Jenkins JN, Saha S (2002) Assessment of day-neutral backcross populations of cotton using AFLP markers. J Cotton Sci 6:97–103
Author information
Authors and Affiliations
Corresponding author
Additional information
Contribution of USDA-ARS in cooperation with the Mississippi Agric. and Forestry Exp. Stn. Journal paper J-11131 of Mississippi Agric. and Forestry Exp. Stn. Mention of trademark, proprietary product, or vendor does not constitute a guarantee or warranty of the product by USDA, ARS and does not imply its approval to the exclusion of other products or vendors that may also be suitable.
Rights and permissions
About this article
Cite this article
Guo, Y., McCarty, J.C., Jenkins, J.N. et al. QTLs for node of first fruiting branch in a cross of an upland cotton, Gossypium hirsutum L., cultivar with primitive accession Texas 701. Euphytica 163, 113–122 (2008). https://doi.org/10.1007/s10681-007-9613-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-007-9613-1