
Summary
Background Adrenal cortical carcinoma (ACC) is a rare cancer with treatment options of limited efficacy, and poor prognosis if metastatic. AT-101 is a more potent inhibitor of B cell lymphoma 2 family apoptosis-related proteins than its racemic form, gossypol, which showed preliminary clinical activity in ACC. We thus evaluated the efficacy of AT-101 in patients with advanced ACC. Methods Patients with histologically confirmed metastatic, recurrent, or primarily unresectable ACC were treated with AT-101 (20 mg/day orally, 21 days out of 28-day cycles) until disease progression and/or prohibitive toxicity. The primary endpoint was objective response rate, wherein a Response Evaluation Criteria In Solid Tumors (RECIST) partial response rate of 25% would be considered promising and 10% not, with a Type I error of 10% and 90% power. In a 2-stage design, 2 responses were required of the first 21 assessable subjects to warrant complete accrual of 44 patients. Secondary endpoints included safety, progression-free survival and overall survival. Results This study accrued 29 patients between 2009 and 2011; median number of cycles was 2. Seven percent experienced grade 4 toxicity including cardiac troponin elevations and hypokalemia. None of the first 21 patients attained RECIST partial response; accordingly, study therapy was deemed ineffective and the trial was permanently closed. Conclusions AT-101 had no meaningful clinical activity in this study in patients with advanced ACC, but demonstrated feasibility of prospective therapeutic clinical trials in this rare cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adrenal cortical carcinoma (ACC) is a rare cancer that is best initially managed with surgical resection even when oligometastatic. [1, 2] Although the chances of disease recurrence are attenuated in patients treated with adjuvant mitotane, the majority of patients undergoing curative-intention resection ultimately later develop widespread metastases. [3] Moreover, many patients have either unresectable or widely metastatic disease at the time of diagnosis. In metastatic disease, chemotherapy combining cisplatin, etoposide, and doxorubicin provides the greatest first-line efficacy. [4] However, in a recent phase 3 clinical trial, the objective response rate (ORR) was only 23%, progression-free survival (PFS) was only 5 months, and overall survival 14.8 months, suggesting benefit, but critical need for improved and additional systemic therapeutic approaches. [5] Unfortunately, subsequent lines of therapy evaluated in clinical trials including streptozocin, other chemotherapy regimens, and targeted agents have response rates about 10%, limited durations of response, and do not definitively improve survival. [5,6,7,8,9] Additional systemic approaches are therefore sorely needed to improve a low 5-year survival of 13%. [10, 11]
Dysregulation of apoptosis is crucial to carcinogenesis and cancer progression and associated with resistance to standard therapy. Thus, targeting these pathways is potentially attractive. In particular, the B cell lymphoma 2 (BCL2) family of apoptosis-related genes is often differentially expressed in a wide variety of malignancies. Although this family of genes has not been extensively studied in ACC, [12] Preclinical evidence suggested that the loss of apoptosis regulating gene bax was associated with adrenocortical cancers compared to benign adrenal tumors. [13] Hence, modulation of this apoptotic pathway appeared worthy of pursuit in ACC.
In this context, we became interested in R-(−)-gossypol acetic acid (AT-101), the levorotatory enantiomer of gossypol, a natural substance found in cottonseed oil. AT-101 is a BCL2 homology domain 3 (BH3) mimetic that inhibits the heterodimerization and impairs function of BCL-xL, BCL-2, BCL-w, and myeloid cell leukemia 1 (MCL-1). Its racemic form has been evaluated in several cancer types including glioma, ACC, breast, colorectal and lung cancers. [14,15,16] Treatment has been well tolerated in these studies, and partial responses or stable disease were noted. In the initial phase I study investigating AT-101 in patients with advanced malignancies, dose limiting toxicities included transaminitis, nausea, and vomiting, with recommended phase II dose of 20 mg daily for 21 days of a 28-day cycle. [16, 17] In a study of 21 patients with metastatic adrenal cancer who received oral gossypol after disease progression on chemotherapeutic agents, three of them had partial responses for over a year, and one attained a minor response to allow surgical resection. [15]
AT-101 is 3 to 4 times more potent than its racemic form in in vitro anti-proliferation assays and animal models of human cancer. [18] Despite the limited clinical benefit of AT-101 observed in other cancer types in prostate and small cell lung cancer, [19, 20] given demonstrated activity of gossypol in ACC, we conducted a phase II study of AT-101 to evaluate its clinical activity in patients with advanced ACC.
Patients and methods
Patient selection
Eligible adult patients had histologically or cytologically confirmed recurrent, metastatic, or primary unresectable ACC with measurable disease per the Response Evaluation Criteria in Solid Tumors (RECIST) criteria [21] and were ≥ 4 weeks from completing their last prior therapies. Prior and concomitant use of mitotane and/or ketoconazole were allowed for patients with hormonal excess. No specific prior therapy was required, nor was any prior therapy exclusionary. Patients were required to have an anticipated ≥12-week life expectancy, ECOG performance status of 0 to 2, normal organ and bone marrow function defined as leukocytes ≥3000/mm3, absolute neutrophil count ≥1500/mm3, platelets ≥100,000/mm3, total bilirubin <1.5 mg/dL, AST ≤2.5 upper limit of normal (ULN), ALT ≤2.5 ULN, and serum creatinine ≤1.7 mg/dL (or creatinine clearance ≥40 mL/min). Patients were required to be able to take oral medications and to provide written informed consent. Patients with symptomatic or progressive brain metastases, presence of a second malignancy or uncontrolled inter-current illnesses were exclusionary. The protocol was conducted under the auspices of the Mayo Phase II Consortium, and approved by the Institutional Review Boards of all participating institutions.
Treatment plan
AT-101 (NSC 726190) was supplied by the National Cancer Institute Cancer Therapy Evaluation Program as 10 mg tablets. Patients took two 10-mg tablets (total dose, 20 mg) of AT-101 orally, daily 21 out of 28 days. Treatment cycle was continued until: 1) the cancer was determined amenable to surgical resection after achieving an objective response to therapy; 2) disease progression; 3) development of inter-current illness that prevented further treatment administration; 4) occurrence of severe adverse events; 5) self-elected withdraw from the study. Patients were monitored for cardiac function including 12-lead ECG and serum troponin level at baseline, and serum troponin level only after the last dose of AT-101 in each cycle of treatment. Upon discontinuation of treatment, patients were followed every 3 months for up to 2 years from off-treatment date. In the event of an adverse event at least possibly related to AT-101, the dose of AT-101 was reduced to 10 mg once daily based on predefined criteria for dose modification and treatment delay. Patients who required additional dose reduction were taken off study.
Study design and statistical analyses
A two-stage phase II study design based on the proportion of patients who have an objective response to treatment, was used, also assessing toxicity and survival. The primary efficacy endpoint evaluated in this trial is the objective response rate (ORR), defined as the proportion of patients who achieve a confirmed objective response to treatment, either partial or complete (i.e. PR or CR), defined by the RECIST criteria. Based on prior clinical trials of either single or multiple cytotoxic agents in advanced ACC, a true ORR of 25% or greater was selected as an indicator that this regimen warrants further investigation with a null hypothesis being that the true ORR is at most 10%. The decision criteria used a Fleming multi-stage design with 10% Type I error and 90% power. [22] Stage 1: Enroll 21 patients. If at least 2 of these 21 evaluable patients have a confirmed response that started in the first 4 cycles of therapy, accrual would continue. Otherwise, the trial will be terminated. Stage 2: Enroll an additional 19 patients, for a total of 40 evaluable patients. If at least 7 of these 40 patients are confirmed objective responders, then the treatment regimen would be deemed to warrant further investigation. A safety stopping rule was also operative: if >3 of the first 15 patients or, if at any time after the first 15 patients, at least 20% of all patients developed ≥grade 4 non-hematologic adverse event felt to be at least possibly related to study treatment, accrual to the study will be suspended to allow for investigation and decision on whether the study should be terminated.
All patients meeting the eligibility criteria who had signed a consent form and begun treatment were considered evaluable for response. The proportion of patients who achieved a confirmed partial response (PR) to treatment was estimated by the standard binomial estimator and 90% confidence intervals that take into account the multi-stage nature of this design. Secondary endpoints included toxicities, progression-free survival (PFS), and overall survival (OS). All toxicities were graded based on the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. [23] The distributions of overall survival time and time to progression were estimated using the method of Kaplan-Meier. In addition, the 6-month progression-free rate was evaluated using the 6-month rates and associated confidence intervals. In addition, competing risk analyses could be done to evaluate time to progression, allowing for going on to alternate treatment or death prior to progression as competing risks.
Results
Baseline characteristics and follow-up information
This study accrued 29 of a targeted 44 patients between March 2009 and August 2011 when it was closed at the futility interim analysis due to lack of activity. Patient baseline characteristics are summarized in Table 1. The median number of cycles completed by patients was 2 (range: 1–11). Twenty-two patients (75.9%) were dead from their disease at the time of data lock date on May 16th, 2012. The median time of follow-up for alive patients was 6.4 months (range: 2.2–10.4 months). Reasons for treatment termination for individual patients included refusal of further treatment in 1 patient (3.4%), disease progression in 24 patients (82.8%), clinical (symptomatic) progression and hospice enrollment in 1 patient (3.4%), manufacturer refusing to provide study drug in 1 patient (3.4%), and adverse events in 2 patients (6.9%). Among these 2 patients, one had grade 4 troponin elevation and the other one had grade 3 hypoalbuminemia that did not resolve within 2 weeks to grade 1 or less.
Treatment summary
A total of 80 cycles of treatment with AT-101 were completed. Four cycles (5%) of the treatment were delayed in 3 patients due to grade 3 nausea/vomiting, grade 4 hypokalemia, and grade 3 AST/ALT elevation, respectively. Four cycles (5%) of the treatment were dose-reduced in 4 patients due to grade 3 nausea/vomiting, grade 3 hypokalemia, elevated AST/ALT, fatigue, respectively. Prior treatments included mitotane, single agent or combination chemotherapy such as cisplatin/carboplatin plus etoposide and doxorubicin (“EDP”), gemcitabine plus docetaxel, and otherwise.
Outcome measures
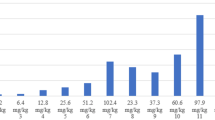
At the time of planned interim analysis, 27 (86.2%) out of 29 evaluable patients had incurred disease progression. No patient attained RECIST PR. Eight patients (27.6%) achieved stable disease for median duration of 3.8 (range 1.8 to 10.1) months. In this group, the change from baseline in the sum of the longest diameter of target lesions varied from −23.9 to +6.6%. One patient (3.4%) only had 1 cycle of treatment without assessment, and thus was unevaluable. Five patients received concomitant mitotane. Among them, three patients had disease progression and two had stable disease as their best response (Fig. 1). Median time to progression was 1.9 months (95% confidence interval: 1.8–2.0 months), and median OS was 8.5 months (95% confidence interval: 5.0–9.8 months), as shown in Fig. 2.

Waterfall plot showing percent radiographic change of tumor at nadir from pretreatment

(a) Time to progression (censored time marked). Median PFS 1.9 months (95% confidence interval: 1.8–2.0 months); (b) Overall survival (censored time marked). Median OS 8.5 months (95% confidence interval: 5.0–9.8 months)
Adverse events
All adverse events regardless of attribution were summarized in Table 2 for a total of 29 evaluable patients. Two patients (12%) experienced grade ≥ 4 non-hematological adverse events including grade 4 cardiac troponin T elevation (probably related) and grade 4 hypokalemia (possibly related).
Discussion
AT-101 as a single agent and as studied in this trial did not show any evidence of clinical activity in patients with advanced ACC. On the surface, this seems quite surprising, given the reported promising clinical activity of its parent racemic mixture, gossypol, in ACC [15] and also given that AT-101 was deemed the more active enantiomer. However, RECIST criteria were not used in the prior study. Instead, response criteria involved 50% estimated reduction in lesion volume. Moreover, some bone lesions were measured in the prior study not deemed measurable per RECIST. [15] Hence, different approaches to assessing response across the two studies perhaps explain the noted discrepancy. It is also possible that co-treatment with mitotane, an inducer of CYP3A4 and an agent that attenuates accumulation of other agents including multikinase inhibitors, might also have lessened the effects of AT-101. It is furthermore possible that targets other than BCl-2 family members may have been invoked by a component of gossypol that was removed in the generation of AT-101, and that the previously posited mechanism was in err, alternatively having potential to explain the present unexpectedly disappointing results. Moreover, the reproducibility of published phase 2 trials on attempted validation is low, generally <50%, [24] also potentially accounting for observed differences. To resolve these differences would require a dedicated confirmatory phase 2 trial of gossypol rather than AT-101 in ACC using RECIST criteria.
The low clinical activity of AT-101 was, however, unfortunately also demonstrated in recent clinical trials evaluating its role in other cancers. For example, phase II studies of AT-101 alone or in combination with topotecan in recurrent and refractory small cell lung cancer did not observe any objective disease responses. [20, 25] AT-101 plus docetaxel was compared with docetaxel alone in a randomized phase II trial as second-line therapy for patients with non-small cell lung cancer, with no ORR or median PFS differences noted between the two arms. [26] AT-101 in combination with standard therapy was also evaluated in phase II trials for both castration-sensitive and castration-resistant metastatic prostate cancer as well as recurrent, advanced head and neck cancer, showing no added clinical benefit with addition of AT-101 to the standard therapy. [27,28,29]
The limited clinical activity of AT-101 as an inhibitor of BCL2 family of apoptosis-related proteins in ACC and other cancer types could be attributed to multiple reasons, but is uncertain. From a pharmacokinetic and pharmacodynamic perspective, lack of sufficient drug concentration in targeted cancer cells at the dosage used in clinical trials often explains why promising findings in vitro often fail to be translated into the clinic, and this might be affected by prior mitotane therapy as one possibility. The concomitant use of mitotane for example, an inducer of CYP3A4, increases the metabolism of some tyrosine kinase inhibitors such as sunitinib and linsitinib (OSI-906), [30] which could explain the negative results from a phase III trial with linsitinib [7] and a phase II trial with sunitinib [8] as well as potentially contribute to the negative results in the present trial. Induced drug resistance could potentially also develop quickly in the presence of redundant BCL-2 family proteins and other pathways involved in cancer cell apoptosis when a target-specific inhibitor is used. In fact, as demonstrated in limited correlative analyses of a phase II trial evaluating AT-101 in small cell lung cancer, AT-101 failed to induce caspase-dependent cell death. [25] Specific to advanced ACC, recent molecular studies revealed several dysregulation of signaling pathways and genomic alterations in advanced ACC including overexpression of insulin-like growth factors 2, β-catenin, VEGFR pathway and mutations in TP53, CKD2A, NF1, RB1 and others rather than BCL2 family proteins. [31,32,33,34] More recent clinical efforts have thus been focusing on evaluating targeted agents on these pathways along with immunotherapeutic strategies. [33]
In summary, AT-101 demonstrated no clinical activity in patients with advanced ACC. This study nevertheless demonstrates the feasibility of conducting prospective therapeutic clinical trials even in this rare tumor. Further drug development in advanced ACC should thus also be feasible but should instead focus on other novel therapeutics.
References
Dy BM, Strajina V, Cayo AK et al (2015) Surgical resection of synchronously metastatic adrenocortical cancer. Ann Surg Oncol 22(1):146–151. https://doi.org/10.1245/s10434-014-3944-7
Dy BM, Wise KB, Richards ML et al (2013) Operative intervention for recurrent adrenocortical cancer. Surgery 154(6):1292–1299; discussion 1299. https://doi.org/10.1016/j.surg.2013.06.033
Terzolo M, Angeli A, Fassnacht M et al (2007) Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 356(23):2372–2380. https://doi.org/10.1056/NEJMoa063360
Berruti A, Terzolo M, Pia A et al (1998) Mitotane associated with etoposide, doxorubicin, and cisplatin in the treatment of advanced adrenocortical carcinoma. Italian Group for the Study of adrenal Cancer. Cancer 83(10):2194–2200
Fassnacht M, Terzolo M, Allolio B et al (2012) Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med 366(23):2189–2197. https://doi.org/10.1056/NEJMoa1200966
Sperone P, Ferrero A, Daffara F et al (2010) Gemcitabine plus metronomic 5-fluorouracil or capecitabine as a second−/third-line chemotherapy in advanced adrenocortical carcinoma: a multicenter phase II study. Endocr Relat Cancer 17(2):445–453. https://doi.org/10.1677/ERC-09-0281
Fassnacht M, Berruti A, Baudin E et al (2015) Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol 16(4):426–435. https://doi.org/10.1016/S1470-2045(15)70081-1
Kroiss M, Quinkler M, Johanssen S et al (2012) Sunitinib in refractory adrenocortical carcinoma: a phase II, single-arm, open-label trial. J Clin Endocrinol Metab 97(10):3495–3503. https://doi.org/10.1210/jc.2012-1419
Ferrari L, Claps M, Grisanti S et al (2016) Systemic therapy in locally advanced or metastatic adrenal cancers: a critical appraisal and clinical trial update. Eur Urol Focus 1(3):298–300. https://doi.org/10.1016/j.euf.2015.06.005
Fassnacht M, Johanssen S, Quinkler M et al (2009) Limited prognostic value of the 2004 International Union against Cancer staging classification for adrenocortical carcinoma: proposal for a revised TNM classification. Cancer 115(2):243–250. https://doi.org/10.1002/cncr.24030
Fassnacht M, Dekkers O, Else T et al (2018) European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European network for the study of adrenal tumors. Eur J Endocrinol 179(4):G1–G46. https://doi.org/10.1530/EJE-18-0608
Kirschner LS (2002) Signaling pathways in adrenocortical cancer. Ann N Y Acad Sci 968:222–239
Kanauchi H, Wada N, Clark OH, Duh QY (2002) Apoptosis regulating genes, bcl-2 and bax, and human telomerase reverse transcriptase messenger RNA expression in adrenal tumors: possible diagnostic and prognostic importance. Surgery 132(6):1021–1026; discussion 1026-7. https://doi.org/10.1067/msy.2002.128616
Bushunow P, Reidenberg MM, Wasenko J et al (1999) Gossypol treatment of recurrent adult malignant gliomas. J Neuro-Oncol 43(1):79–86
Flack MR, Pyle RG, Mullen NM et al (1993) Oral gossypol in the treatment of metastatic adrenal cancer. J Clin Endocrinol Metab 76(4):1019–1024. https://doi.org/10.1210/jcem.76.4.8473376
Stein RC, Joseph AE, Matlin SA et al (1992) A preliminary clinical study of gossypol in advanced human cancer. Cancer Chemother Pharmacol 30(6):480–482
Investigator’s Brochure (2006) AT-101 [R-(−)-gossypol acetic acid]. Ascenta Therapeutics, Inc. Accessed 31 Mar 2006
Shelley MD, Hartley L, Fish RG et al (1999) Stereo-specific cytotoxic effects of gossypol enantiomers and gossypolone in tumour cell lines. Cancer Lett 135(2):171–180
Liu G, Kelly WK, Wilding G et al (2009) An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin Cancer Res 15(9):3172–3176. https://doi.org/10.1158/1078-0432.CCR-08-2985
Heist RS, Fain J, Chinnasami B et al (2010) Phase I/II study of AT-101 with topotecan in relapsed and refractory small cell lung cancer. J Thorac Oncol 5(10):1637–1643. https://doi.org/10.1097/JTO.0b013e3181e8f4dc
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247. https://doi.org/10.1016/j.ejca.2008.10.026
Fleming TR (1982) One-sample multiple testing procedure for phase II clinical trials. Biometrics 38(1):143–151
Trotti A, Colevas AD, Setser A et al (2003) CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol 13(3):176–181. https://doi.org/10.1016/S1053-4296(03)00031-6
Sharma MR, Stadler WM, Ratain MJ (2011) Randomized phase II trials: a long-term investment with promising returns. J Natl Cancer Inst 103(14):1093–1100. https://doi.org/10.1093/jnci/djr218
Baggstrom MQ, Qi Y, Koczywas M et al (2011) A phase II study of AT-101 (gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J Thorac Oncol 6(10):1757–1760. https://doi.org/10.1097/JTO.0b013e31822e2941
Ready N, Karaseva NA, Orlov SV et al (2011) Double-blind, placebo-controlled, randomized phase 2 study of the proapoptotic agent AT-101 plus docetaxel, in second-line non-small cell lung cancer. J Thorac Oncol 6(4):781–785. https://doi.org/10.1097/JTO.0b013e31820a0ea6
Sonpavde G, Matveev V, Burke JM et al (2012) Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Ann Oncol 23(7):1803–1808. https://doi.org/10.1093/annonc/mdr555
Stein MN, Hussain M, Stadler WM et al (2016) A phase II study of AT-101 to overcome Bcl-2--mediated resistance to androgen deprivation therapy in patients with newly diagnosed castration-sensitive metastatic prostate Cancer. Clin Genitourin Cancer 14(1):22–27. https://doi.org/10.1016/j.clgc.2015.09.010
Swiecicki PL, Bellile E, Sacco AG et al (2016) A phase II trial of the BCL-2 homolog domain 3 mimetic AT-101 in combination with docetaxel for recurrent, locally advanced, or metastatic head and neck cancer. Investig New Drugs 34(4):481–489. https://doi.org/10.1007/s10637-016-0364-5
Kroiss M, Quinkler M, Lutz WK et al (2011) Drug interactions with mitotane by induction of CYP3A4 metabolism in the clinical management of adrenocortical carcinoma. Clin Endocrinol 75(5):585–591. https://doi.org/10.1111/j.1365-2265.2011.04214.x
Almeida MQ, Fragoso MCBV, Lotfi CFP et al (2008) Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. J Clin Endocrinol Metab 93(9):3524–3531. https://doi.org/10.1210/jc.2008-0065
Assié G, Letouzé E, Fassnacht M et al (2014) Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46(6):607–612. https://doi.org/10.1038/ng.2953
Varghese J, Habra MA (2017) Update on adrenocortical carcinoma management and future directions. Curr Opin Endocrinol Diabetes Obes 24(3):208–214. https://doi.org/10.1097/MED.0000000000000332
Zheng S, Cherniack AD, Dewal N et al (2016) Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 29(5):723–736. https://doi.org/10.1016/j.ccell.2016.04.002
Funding
This study is supported by The Phase 2 Consortium (P2C) through its contract with the National Cancer Institute (N01 CM17104).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Hao Xie declares that he has no conflict of interest. Jun Yin declares that she has no conflict of interest. Manisha H. Shah declares that she has no conflict of interest related to AT-101 except research funding from Bristol-Myers Squibb for a clinical trial in last 2 years. Michael E. Menefee declares that he has no conflict of interest. Keith C. Bible declares that he has no conflict of interest. Diane Reidy-Lagunes declares that she receives research funds from Novartis, Ipsen, and Merck, and is on the advisory board for Novartis, Ipsen, AAA, and Lexicon. Madeleine A. Kane declares that she has no conflict of interest. David I. Quinn declares that he has no conflict of interest. David R. Gandara declares that he has no conflict of interest. Charles Erlichman declares that he has no conflict of interest. Alex A. Adjei declares that he has no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xie, H., Yin, J., Shah, M.H. et al. A phase II study of the orally administered negative enantiomer of gossypol (AT-101), a BH3 mimetic, in patients with advanced adrenal cortical carcinoma. Invest New Drugs 37, 755–762 (2019). https://doi.org/10.1007/s10637-019-00797-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-019-00797-1