
Summary
Background The MAPK pathway plays a central role in regulation of several cellular processes, and its dysregulation is a hallmark of biliary tract cancer (BTC). Binimetinib (MEK162), a potent, selective oral MEK1/2 inhibitor, was assessed in patients with advanced BTC. Patients and Methods An expansion cohort study in patients who received ≤1 line of therapy for advanced BTC was conducted after determination of the maximum tolerated dose in this Phase 1 trial. Patients received binimetinib 60 mg twice daily. The primary objectives were to characterize the safety profile and pharmacokinetics of binimetinib in advanced BTC. Secondary objectives included assessment of clinical efficacy, changes in weight and lean body mass, and pharmacodynamic effects. Tumor samples were assessed for mutations in relevant genes. Results Twenty-eight patients received binimetinib. Common adverse events (AEs) were mild, with rash (82%) and nausea (54%) being most common. Two patients experienced grade 4 AEs, one generalized edema and the other pulmonary embolism. The pharmacokinetics in this patient population were consistent with those previously reported (Bendell JC et al., Br J Cancer 2017;116:575-583). Twelve patients (43%) experienced stable disease and two had objective responses (1 complete response, 1 partial response) per Response Evaluation Criteria in Solid Tumors and stable metabolic disease by positron emission tomography/computed tomography. Most patients (18/25; 72%) did not have KRAS, BRAF, NRAS, PI3KCA, or PTEN mutations, nor was there correlation between mutation status and response. The average non-fluid weight gain was 1.3% for lean muscle and 4.7% for adipose tissue. Conclusion Binimetinib was well tolerated and showed promising evidence of activity in patients with BTC. Correlative studies suggested the potential for binimetinib to promote muscle gain in patients with BTC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Biliary tract cancer (BTC) arises from the epithelium of the intrahepatic and extrahepatic biliary tree [1]. Despite modest treatment advances, BTC has a poor prognosis, with a median survival of <1 year [1]. The current standard regimen for treatment of advanced BTC is gemcitabine and cisplatin. Results from clinical trials for refractory BTC have been disappointing, highlighting the need for more effective therapies [2].
The mechanisms of cholangiocarcinogenesis are complex, involving multiple molecular signaling pathways and inflammatory cytokines [3, 4]. Inappropriate activation of the RAS/RAF/MEK/ERK (MAPK) pathway in BTC appears to be common and can occur through several distinct mechanisms, including mutations in RAS, BRAF, [5], and growth factor signaling [6, 7], as well as the release of cytokines [8]. Inhibition of the MAPK pathway may impede cancer signaling through multiple mechanisms, making it an attractive target for treatment of BTC. Anti-tumor activity has been observed with the MEK1/2 inhibitor selumetinib in patients with BTC [9], as well as the ability to increase lean muscle weight [10].
Binimetinib (MEK162) is a potent, selective, allosteric, ATP-uncompetitive inhibitor of MEK1/2 that reduces ERK phosphorylation and growth of BRAF- or KRAS-mutant cancer cells at low nanomolar ranges. In a dose-escalation phase 1 study in patients with advanced solid tumors, the single-agent maximum tolerated dose (MTD) of binimetinib was determined to be 60 mg orally twice daily (BID) [11]. Here we present data from an expansion cohort in patients with advanced BTC. We further characterize safety and pharmacokinetic (PK) profiles, preliminary efficacy, effects on cachexia, and exploratory biomarkers of binimetinib treatment in this population.
Patients and methods
This study (ClinicalTrials.gov, NCT00959127) was conducted under all applicable regulatory requirements with approval by institutional review boards of all participating sites. Patients provided written informed consent before initiation of study-related procedures.
Study design
This was a multi-center, open-label phase 1 study that included a dose-escalation portion followed by an expansion cohort. Following the determination of MTD of binimetinib 60 mg orally BID, three expansion cohorts were enrolled. Two of the cohorts comprised patients with KRAS-mutant colorectal cancer and BRAF-mutant colorectal cancer. Primary objectives of the third expansion cohort were to further characterize the safety profile and PK of binimetinib in patients with advanced or metastatic BTC (intrahepatic and extrahepatic cholangiocarcinoma [ICC/ECC] or gallbladder carcinoma [GBC]). Secondary objectives included obtaining preliminary estimates of efficacy, assessing changes in weight and lean body mass, determining mutation status from tumor biopsy samples, and assessing pharmacodynamic (PD) effects.
Binimetinib administration
Patients received binimetinib 60 mg BID in continuous 21-day cycles, and continued unless they withdrew informed consent or experienced disease progression or unacceptable toxicity. Binimetinib dose was permitted to be reduced to successive levels of 45 mg BID, 30 mg BID, and 20 mg BID or interrupted, as appropriate, based on protocol-defined treatment modifications.
Patient population
Patients were ≥18 years of age with histologically or cytologically confirmed BTC that was unresectable, locally advanced, or metastatic, and with either measurable or evaluable non-measurable disease. Patients were required to have received no more than 1 prior anti-cancer therapy (including adjuvant therapy), an Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0 or 1, and a cardiac ejection fraction as measured by left ventricular ejection fraction (LVEF) greater than or equal to the institutional lower limit of normal, as well as adequate bone marrow, renal and hepatic function, and archival tissue or a fresh biopsy submitted for PD analysis. Patients were excluded if they had a history of central serous retinopathy (CSR), baseline risk factors for CSR, retinal vein occlusion, or if they had received previous treatment with a MEK inhibitor.
Safety assessments
Assessments included adverse event (AE) monitoring, standard laboratory parameters, electrocardiograms, ECOG PS, vital signs measurements, physical examinations, ophthalmologic examinations, and LVEF. AEs were coded using the Medical Dictionary for Regulatory Activities, version 12.0. AE severity was assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0. All patients who received ≥1 dose of binimetinib were included in the safety analysis.
Efficacy assessments
Efficacy assessments included analyses of tumors by radiologic measures, serologic tumor markers, and survival data. Computed tomography (CT) was performed every 6 weeks and evaluated by the investigator using Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1 [12]. Duration of response was calculated for patients who achieved a confirmed complete response (CR) or partial response (PR). Kaplan-Meier estimates were performed for progression-free survival (PFS) and overall survival (OS) [13]. Patients in the safety population who had ≥1 measurable lesion at baseline and ≥1 post-baseline disease assessment were evaluated for efficacy.
Correlative studies
Lean body mass changes
An independent clinician assessed lean body mass at baseline and subsequent time points using the CT scans. Skeletal muscle cross-sectional area (cm2) was calculated at the third lumbar vertebra (L3) as previously described [14, 15].
Lean muscle assessment was conducted only while patients were receiving binimetinib and results were presented as change per 100 days. In a post-hoc analysis, changes in muscle and fat areas were presented as the maximum percentage change from baseline in cm2 lost or gained/number of days between scans × 100 to account for between-patient variations in timing of post-baseline scans. In a further post-hoc analysis, patients were analyzed by categories of significant muscle gain, significant muscle loss, or stable muscle mass, defined as a change of 1 kg of skeletal muscle [16].
Pharmacokinetic analysis
Venous blood samples were drawn for measurement of plasma binimetinib concentrations by both intensive and more limited PK sampling (see Supplementary Materials for time points). Standard non-compartmental PK parameters were calculated on serial PK collection days (Cycle 1 Day 15 and Cycle 3 Day 1) for each patient and then summarized.
Biomarker analysis
Formalin-fixed/paraffin-embedded or fresh frozen tissue samples were obtained from all patients before treatment. KRAS, NRAS, BRAF, and PI3KCA mutations were assessed using the Sequenom OncoCarta Assay Panel (Sequenom, San Diego, CA) and/or BEAMing Digital PCR (Inostics GmbH, Hamburg, Germany) according to the manufacturers’ instructions. PTEN expression was determined by immunohistochemistry reported as an H-score and classified as PTEN negative (H-score < 50) or PTEN positive (H-score ≥ 50).
Pharmacodynamic analysis
Pre-dose venous blood samples were collected at baseline, Day 8 of Cycle 1, Day 15 of Cycle 1, and Day 1 of all subsequent cycles for measurement of tumor necrosis factor alpha (TNF-α), interferon (IFN), interleukin (IL)-10, IL-12p70, IL-1β, IL-6, and IL-8 using Meso Scale Discovery Human ProInflammatory 7-Plex Ultra-Sensitive Assay kit (K15008C; Meso Scale Diagnostics, LLC, Gaithersburg, MD) according to the manufacturers’ instructions. C-reactive protein (CRP) was analyzed using Meso Scale Discovery Human CRP Assay kit (K151EPC; Meso Scale Diagnostics, LLC).
Results
Twenty-eight patients were enrolled between April and September 2010. Median patient age was 64 years, and 16 patients (57%) were female. Fourteen (50%) had ICC, 7 (25%) had ECC, and 7 (25%) had GBC (Table 1). The majority had an ECOG PS of 1 (64%). Twelve patients (43%) received first-line systemic anti-cancer therapy, 5 (18%) received only adjuvant therapy, and 11 (39%) received no prior chemotherapy.
Safety and tolerability
The most common AEs (all grades, regardless of causality) were rash, nausea, vomiting, diarrhea, fatigue, and peripheral edema (Table 2). Toxicities were mostly grade 1 or 2 and reversible. Two patients experienced a grade 4 AE, one due to generalized edema and the other due to pulmonary embolism. No grade 5 events were reported. Laboratory abnormalities included hyponatremia and elevations in creatine phosphokinase and liver function tests (Supplementary Table 1).
Five patients (18%) discontinued binimetinib because of an AE, 3 (11%) of whom did so because of AEs considered to be related to binimetinib, including chorioretinopathy, generalized edema, and nausea in 1 patient each (4%). Thirteen patients (46%) required a dose reduction, most commonly because of eye disorders of retinal “non-tear” detachment (3 patients, 11%), and chorioretinopathy and macular edema (2 patients each, 7%). All ocular toxicities were diagnosed by optical coherence tomography examinations and were reversible upon binimetinib dose reduction.
Twelve patients (43%) reported serious adverse events (SAEs) during the study or within 30 days of the last binimetinib dose, and among them 4 (14%) had at least 1 SAE that was considered related to binimetinib, specifically generalized edema, mucosal inflammation, retinal detachment, and upper and lower gastrointestinal hemorrhage in 1 patient each (4%). Ten patients (36%) died while on study drug or within 30 days of the last dose, and the cause of all deaths was disease progression.
Efficacy
Twenty-six patients (93%) were evaluable for efficacy response. Among them, 2 patients (8%) had an objective response, including 1 CR of 11.3 months duration and 1 PR of 17.9 months. Neither of those responders had detectable mutations in KRAS, BRAF, NRAS, PI3KCA, and PTEN.
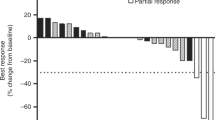
Twelve patients (43%) experienced stable disease (SD), ranging from 0.9 to 7.3 months. Figure 1a illustrates the change in the sum of longest diameters of target lesions in evaluable patients with measurable disease. Median PFS was 2.1 months (95% CI, 1.4–4.8) and median OS was 4.8 months (95% CI, 3.5–10.0; Fig. 1b).

a Waterfall plot of maximum percentage change in target lesions among patients with measurable disease evaluable for response (N = 21). b Kaplan-Meier plot for PFS and OS. CR, complete response; EH, extrahepatic; GB, gallbladder; IH, intrahepatic; Max, maximum; OS, overall survival; PD, progressive disease; PFS, progression-free survival; PR, partial response; SD, stable disease.*Patient remained on binimetinib at the time of maximal response. Note: open circles represent patients with censored time points
Thirteen patients (46%) had 2-[F-18]fluoro-2-deoxy-D-glucose-PET results assessed quantitatively by standardized uptake values. Of these, 4 (31%) had a metabolic PR, 3 (23%) had metabolic SD (including 2 patients with RECIST responses), and 2 (15%) had progressive metabolic disease. The remaining 4 patients did not have post-baseline measurements.
Mutational analysis
All evaluated tumor samples were archived histological specimens (n = 25). Eighteen patients (64%) had no mutations detected in KRAS, BRAF, NRAS, PI3KCA, and PTEN. Four (14%) had unknown mutational status, 2 (7%) had KRAS mutations, 2 (7%) had PTEN mutations, 1 (4%) had a PI3KCA mutation, and 1 (4%) had c-MET amplification. There was no correlation between objective response and mutational status.
Weight, lean muscle, and subcutaneous fat changes
Change in lean body mass was assessed in 17 patients (61%). Mean maximal changes from baseline in muscle and adipose tissues were 0.9% (range, −11.1% to 11.8%) and 4.2% (range, −34.4% to 54.1%), respectively. The median maximum change in body weight was 3.3% (range, −5.0% to 15.5%). After adjusting for variations between patients in time intervals between baseline and follow-up CT scans, the mean change (standard deviation) in muscle surface area/100 days was 1.64 cm2 (35.89 cm2). Among the 20 patients from whom data were available, 7 (35%) gained >1 kg of muscle, 9 (45%) lost >1 kg of muscle, and 4 (20%) had stable muscle mass (Supplementary Figure 1).
Pharmacokinetics
In the intensive PK sampling group (n = 7), geometric mean area under the curve (AUC) from time 0 to 8 h (AUC0–8) and maximum serum concentration (Cmax) values were 1090 h∙ng/mL and 365 ng/mL, respectively, on Cycle 1 Day 1 and 3760 h∙ng/mL and 594 ng/mL on Cycle 1 Day 15. The calculable accumulation ratio (RAUC) was 2.5, indicating a modest accumulation of binimetinib after daily BID dosing.
On Cycle 1 Day 15 in the limited PK sampling group (n = 20), the geometric mean AUC0–8 value was 2090 h·ng/mL and the geometric mean Cmax value was 497 ng/mL. On Cycle 3 Day 1, the geometric mean binimetinib AUC0–8 was 1530 h·ng/mL and the geometric mean Cmax was 540 ng/mL.
Pharmacodynamics
Review of individual patient data for CRP, IFN, IL-10, IL-12p70, IL-1β, IL-6, and IL-8 serum concentrations indicated no notable changes relative to baseline following binimetinib treatment. Serum concentrations of TNF-α were evaluated pre-dose on Cycle 1 Days 8 and 15. Median decreases of TNF-α were similar at both time points and ranged from 34% to 35% of baseline.
Discussion
Despite treatment advances, prognosis for patients with BTC remains poor. In this BTC expansion cohort study binimetinib was administered safely, with manageable dermatologic and gastrointestinal toxicities. All toxicities were reversible, including ocular toxicities, and were consistent with known class effects of MEK inhibition. In the extended PK sampling scheme, modest accumulation of binimetinib occurred as indicated by an RAUC of 2.5; overall however, binimetinib exposure in terms of both AUC0-8 and Cmax was within the ranges previously reported [11].
Clinical responses included durable CR and PR. Expanded mutational profile testing did not identify a mutational status and response relationship. Recent studies have identified signatures that may predict response to MEK inhibition without mutations in the MAPK pathway [17].
Other mechanisms may explain the anti-tumor activity of binimetinib in BTC, including potential immunomodulatory effects [18]. Inflammatory cytokines cause the proliferation of biliary cancer cells and activation of cell-survival pathways, including RAS/RAF/MEK/ERK [19,20,21]. Although the findings of the current study did not show a clear correlation between treatment and changes in cytokine levels, MEK inhibition may alter immune cell function and phenotype in BTC [21].
Patients experienced non-fluid increases in weight, with an average 0.9% lean muscle and 4.2% adipose tissue gain. This study confirms prior results with another MEK inhibitor, selumetinib [10]. The findings of the current study may be explained by the immunomodulatory effects of binimetinib on TNF-α levels. However, there were no notable changes in other cytokines that have also been implicated in cancer-associated cachexia [22,23,24].
In conclusion, treatment with binimetinib was well tolerated and showed evidence of activity in BTC. More work is needed to better understand the underlying mechanisms of activity of the drug and how to select patients who will respond optimally to treatment. Other studies are under way with binimetinib and other MEK inhibitors as single agents or in combination in BTC.
References
Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD (2005) Cholangiocarcinoma. Lancet 366:1303–1314
Sadeghi S, Finn RS (2014) Systemic therapy for cholangiocarcinoma. Clin Liver Dis 3:86–89
Goydos JS, Brumfield AM, Frezza E et al (1998) Marked elevation of serum interleukin-6 in patients with cholangiocarcinoma: validation of utility as a clinical marker. Ann Surg 227:398–404
Park J, Tadlock L, Gores GJ, Patel T (1999) Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology 30:1128–1133
Davies H, Bignell GR, Cox C et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954
Nakazawa K, Dobashi Y, Suzuki S et al (2005) Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J Pathol 206:356–365
Bennasroune A, Gardin A, Aunis D et al (2004) Tyrosine kinase receptors as attractive targets of cancer therapy. Crit Rev Oncol Hematol 50:23–38
McCubrey JA, May WS, Duronio V, Mufson A (2000) Serine/threonine phosphorylation in cytokine signal transduction. Leukemia 14:9–21
Bekaii-Saab T, Phelps MA, Li X et al (2011) Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol 29:2357–2363
Prado CM, Bekaii-Saab T, Doyle LA et al (2012) Skeletal muscle anabolism is a side effect of therapy with the MEK inhibitor: selumetinib in patients with cholangiocarcinoma. Br J Cancer 106:1583–1586
Bendell JC, Javle M, Bekaii-Saab TS et al (2017) A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Cancer 116:575–583
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Kaplan E, Meier P (1958) Nonparametric estimation from incomplete observations. J Am Stat Assoc 53:457–481
Shen W, Punyanitya M, Wang Z et al (2004) Total body skeletal muscle and adipose tissue volumes: estimation from a single abdominal cross-sectional image. J Appl Physiol (1985) 97:2333–2338
Prado CM, Birdsell LA, Baracos VE (2009) The emerging role of computerized tomography in assessing cancer cachexia. Curr Opin Support Palliat Care 3:269–275
Prado CM, Sawyer MB, Ghosh S et al (2013) Central tenet of cancer cachexia therapy: do patients with advanced cancer have exploitable anabolic potential? Am J Clin Nutr 98:1012–1019
Dry JR, Pavey S, Pratilas CA et al (2010) Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res 70:2264–2273
Gores GJ (2003) Cholangiocarcinoma: current concepts and insights. Hepatology 37:961–969
Mon NN, Kokuryo T, Hamaguchi M (2009) Inflammation and tumor progression: a lesson from TNF-alpha-dependent FAK signaling in cholangiocarcinoma. Methods Mol Biol 512:279–293
Okada K, Shimizu Y, Nambu S et al (1994) Interleukin-6 functions as an autocrine growth factor in a cholangiocarcinoma cell line. J Gastroenterol Hepatol 9:462–467
Maemura K, Natsugoe S, Takao S (2014) Molecular mechanism of cholangiocarcinoma carcinogenesis. J Hepatobiliary Pancreat Sci 21:754–760
Tai YT, Fulciniti M, Hideshima T et al (2007) Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood 110:1656–1663
Zhang D, Zheng H, Zhou Y et al (2007) Association of IL-1beta gene polymorphism with cachexia from locally advanced gastric cancer. BMC Cancer 7:45
Zhang D, Zhou Y, Wu L et al (2008) Association of IL-6 gene polymorphisms with cachexia susceptibility and survival time of patients with pancreatic cancer. Ann Clin Lab Sci 38:113–119
Funding
This work was supported by Array BioPharma Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
Dr. R. S. Finn is a consultant for Bayer, Novartis, Pfizer Inc., Bristol-Myers Squibb. Dr. D. H. Ahn is a consultant for Merrimack. Dr. A. Patnaik receives institutional research funding from Array BioPharma Inc. R. Chavira, J. Christy-Bittel, and Dr. Barrett are current or former employees, and may own stock in, Array BioPharma Inc. All remaining authors have declared no conflicts of interest.
Additional information
Key message: MAPK pathway dysregulation is common in biliary tract cancer and is a key mechanism for tumor proliferation and treatment resistance. Its inhibition represents a novel, relevant treatment strategy in this disease. Binimetinib was well tolerated and showed promising clinical activity. Future studies assessing biomarkers will help determine patients that are likely to benefit from binimetinib.
Electronic supplementary material
ESM 1
(PDF 101 kb)
Rights and permissions
About this article

Cite this article
Finn, R.S., Ahn, D.H., Javle, M.M. et al. Phase 1b investigation of the MEK inhibitor binimetinib in patients with advanced or metastatic biliary tract cancer. Invest New Drugs 36, 1037–1043 (2018). https://doi.org/10.1007/s10637-018-0600-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-018-0600-2