
Summary
Apoptin is a nonstructural protein encoded by one of the three open reading frames of the chicken anemia virus genome. It has attracted a great deal of interest due to its ability to induce apoptosis in multiple transformed and malignant mammalian cell lines without affecting primary and non-transformed cells. However, the use of Apoptin as an anticancer drug is restricted by its strong tendency to aggregate. A number of methods to overcome this problem have been proposed, including transduction techniques to deliver the Apoptin gene into tumor cells, but all such methods have certain drawbacks. Here we describe that a truncated variant of Apoptin, lacking residues 1 to 43, is a soluble, non-aggregating protein that maintains most of the biological properties of wild-type Apoptin when transfected into cells. We show that the cytotoxic effect of this variant is also present when it is added exogenously to cancer cells, but not to normal cells. In addition to the interest this protein has attracted as a promising therapeutic strategy, it is also an excellent model to study the structural properties of Apoptin and how they relate to its mechanism of action.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Apoptin gene is one of the three open reading frames of the chicken anemia virus genome and codes for a protein that is essential for the replication of this virus [1]. What makes Apoptin so interesting is its ability to induce apoptosis in multiple transformed and malignant cell lines while leaving primary and non-transformed cells unaffected [2, 3]. When Apoptin is expressed in living cells, it forms large aggregated complexes [4] that bind to DNA [5], although it does not act as a transcriptional repressor or activator [6]. Apoptin aggregates produced in vitro are toxic to cancer cells when they are microinjected into them, but not to normal cells [4]. Apoptin has also shown to be effective in reducing the size of xenografted tumors in mice using various delivery strategies (for a review, see [7]).
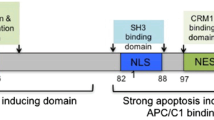
The selective cytotoxicity of Apoptin has been attributed to its differential subcellular localization in tumor and normal cells. At its C-terminus, Apoptin contains a bipartite nuclear localization sequence (NLS1 and NLS2) and a nuclear export sequence (NES) which is sandwiched between NLS1 and NLS2 (see supplementary Fig. 1S). Adjacent to the NES lies Thr108, whose phosphorylation prevents NES recognition [8, 9]. Phosphorylation of Thr108 is catalyzed by tumor-specific kinases and therefore, in tumor cells, Apoptin mainly accumulates in the nucleus whereas in normal cells it is mainly localized in the cytoplasm, where it is degraded. Some studies have indicated that non-phosphorylatable Apoptin mutants can still induce apoptosis of cancer cells (for a review, see [10]). This has been attributed to the existence – within the N-terminus – of a second apoptosis motif whose action is phosphorylation-independent [6]. The N-terminal region of Apoptin possesses a Proline-rich segment (PRS) which spans residues 8 to 28 and a Leucine-rich segment (LRS) which spans residues 33 to 46 and which promotes aggregation [4]. Both the N- and C-terminal regions have inherent cell-killing activity when transfected into cancer cells, although they are less effective than the full-length protein. Interestingly, both regions are able to bind dsDNA [11].
Effective delivery of Apoptin into tumor cells remains a challenge because this protein is highly prone to aggregation. Transduction tools have been used to efficiently deliver the Apoptin gene into tumor cells (for a review, see [7]). However, the therapeutic applications of such approaches have met with certain difficulties including insertional mutagenesis in transfected cells or transient expression. Here, we describe the construction of a truncated variant of Apoptin, which behaves as a soluble, non-aggregation prone protein. This variant is cytotoxic for tumor cells but leaves normal cells unaffected and this cytotoxic effect is produced both when the variant is transfected into cancer cells as well as when it is added exogenously to them.
Experimental procedures
Plasmid construction
The Apoptin gene, cloned into plasmid pcDNA3.1 [12], was generously provided by Dr. Malvash Tavassoli (King’s College, UK). The full-length Apoptin gene was subcloned into pET-28a (Novagen, Germany), resulting in plasmid pET-28Apop. This construction has an N-terminal hexahistidine tag (H6-Apoptin). H6-ApopΔPro and H6-ApopΔProΔLeu (Supplementary Fig. 1S) were constructed from pET-28Apop by introducing a second NdeI restriction site within the H6-Apoptin gene at codon 27 and codon 43, respectively. The resulting mutated variants were digested with NdeI and subsequently ligated to create pET-28ApopΔPro and pET-28ApopΔProΔLeu. To obtain GFP variants, the GFP gene from plasmid pRSET/EmGFP (Invitrogen, USA) was amplified by PCR and introduced into pcDNA3.1 to create pcDNA3.1GFP. To obtain the GFP-Apoptin variants, the GFP and the Apoptin genes were excised and cloned together into pcDNA3.1 resulting in plasmids pcDNA3.1Apop, pcDNA3.1ApopΔPro, and pcDNA3.1ApopΔProΔLeu (Supplementary Fig. 1S). These constructions were confirmed by DNA sequencing.
Protein expression and purification
The Apoptin variants were produced from E. coli BL21 (DE3) transformed cells. Cells were lysed using a French Press (SLM-Aminco, UK); inclusion bodies were harvested by centrifugation and solubilized in 50 mM Tris-HCl, 500 mM NaCl, 7 M Urea pH 7.4. Reduced glutathione (GSH) was added to get a final concentration of 100 mM; pH was adjusted to 7.4 and the sample was incubated at room temperature for 2 h under N2 atmosphere. The sample was incubated overnight with Ni-NTA agarose (Qiagen, Netherlands). The resin was loaded onto a column, washed, and eluted with buffer containing 50 mM Tris-HCl, 500 mM NaCl, 7 M Urea, and 500 mM imidazole pH 7.4. Those samples carrying the desired protein were gathered and diluted dropwise, 1:10, into an aqueous solution containing 50 mM Tris-HCl, 200 mM NaCl, 100 mM L-Arg, 100 mM L-Glu, 1 mM oxidized glutathione, 1 mM GSH, 1 mM EDTA, pH 8.1, and incubated for 24 h at 4 °C. The sample was concentrated by tangential ultrafiltration, dialyzed against ultrapure water and lyophilized. The Apoptin variants were further purified on a Superdex-75 10/300GL (GE Healthcare, UK) column. Purity was analyzed by SDS-PAGE (Supplementary Fig. 2S) and the expected molecular mass was confirmed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. The yield of pure protein per liter of induced culture was approximately 20 mg. The molar extinction coefficients of the variants were 1989 M−1·cm−1 for H6-Apoptin; 1856 M−1·cm−1 for H6-ApopΔPro, and 3775 M−1·cm−1 for H6-ApopΔProΔLeu.
Cell lines and culture conditions
NCI-H460 human lung cancer and OVCAR-8 human ovarian cancer cell lines were obtained from the DCTD tumor/cell line repository of the National Cancer Institute (Frederick, Maryland USA). CCD-18Co human colon fibroblasts and Jurkat human T-cell lymphocyte cell line were acquired from Celltec UB (Universitat de Barcelona, Spain). 1BR.3.G human skin fibroblast cells were obtained from the European Collection of Cell Cultures (Porton Down, UK). K-562 human chronic myelogenous leukemia cells were generously provided by Dr. Bruno Beaumelle (CPBS, Montpellier France).
The NCI-H460, OVCAR-8, K-562 and Jurkat cells were routinely grown in RPMI, supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin, 50 μg/ml streptomycin and 2 mM L-glutamine. The CCD-18Co and 1BR.3.G cells were grown in DMEM, supplemented with 10% FBS, 50 U/ ml penicillin, 50 μg/ml streptomycin and 2 mM L-glutamine. In all cases, the cells were used immediately after resuscitation and were maintained at 37 °C in a humidified atmosphere with 5% CO2, propagated following established protocols, remaining free of mycoplasma throughout the experiments.
Transfection assays
7 × 104 1BR.3.G and 3.5 × 104 NCI-H460 cells were seeded in 24-well plates and transfected after 24 h with 0.5 μg DNA using Lipofectamine® 3000 (Invitrogen, USA) according to the specifications of the manufacturer. 1.5 × 105 CCD-18Co cells were seeded in 6-well plates and transfected after 24 h with 2.5 μg DNA following the protocol described in [13].
Cell proliferation assays
To investigate the effect of the transfection of Apoptin variants, NCI-H460 and 1BR.3.G cells were seeded and transfected as described above. After 48 h of incubation, attached and floating cells were harvested by centrifugation, washed twice in cold PBS and subjected to propidium iodide (PI) staining for 15 min in the dark. Stained cells expressing GFP or GFP-fused proteins were analyzed on a FACSCalibur flow cytometer (BD Biosciences, US) using CellQuest Pro software. A minimum of 10,000 cells within the gated region were analyzed.
To investigate the effect of exogenously added H6-Apoptin and H6-ApopΔProΔLeu protein variants, cells were seeded into 96-well plates at the appropriate density. After 24 h of incubation, cells were treated with various concentrations of H6-Apoptin or H6-ApopΔProΔLeu for 72 h. Cell viability was determined by the MTT method (Sigma, USA) as described elsewhere [14]. The IC50 value represents the concentration of the assayed protein required to inhibit cell proliferation by 50%, in comparison with untreated cells. All data are reported as the mean ± SE of at least three independent experiments, each with three replicates.
Apoptosis assays
Caspase-3, −8 and −9 catalytic activities were measured using the APOPCYTO Caspase-3, −8 and −9 colorimetric assay kits (MBL, Japan) according to the specifications of the manufacturer. Quantitative analysis of NCI-H460 apoptotic cell death caused by treatment with 15 μM H6-ApopΔProΔLeu was performed by flow cytometry using the Alexa Fluor® 488 annexin V/PI Vybrant Apoptosis Assay Kit (Molecular Probes, USA) as described elsewhere [15]. All the results are reported as the mean ± SE of at least three independent experiments.
Transmission electron microscopy
H6-Apoptin was prepared at a concentration of 1 mg/ml and incubated for 72 h at 37 °C in 50 mM Tris-HCl pH 7.0. Negatively stained samples were prepared on carbon-coated grids with 2% (w/v) uranyl acetate solution [16]. TEM images were obtained using a Zeiss EM910 microscope at 60 kV at the Technical Research Services of the Universitat de Girona.
Dynamic light scattering
Dynamic light scattering (DLS) measurements were recorded on a Malvern ZetaSizer Nano ZS (532 nm) at 25 °C in PBS at a final protein concentration of 1 mg/ml at the Scientific and Technical Services of the Universitat Autònoma de Barcelona. The hydrodynamic radius of the particle (RH) was determined by averaging the results of at least three independent experiments. An upper estimate of the protein molecular mass was obtained from the RH value using the empirical relationship for typical globular proteins: MM (kDa) = (1.68xRH)2.34 using the supplied software.
Confocal microscopy
NCI-H460 and CCD-18Co cells were seeded on coverslips and allowed to attach overnight after which they were transfected and incubated at 37 °C and 5% CO2 for 24 h. They were then fixed and the nuclei stained with 2 μg/ml Hoechst 33258 for 15 min at room temperature. For intracellular localization studies, samples were washed twice with cold PBS, whereas for the study of apoptotic activity the coverslips were directly mounted. All samples were examined using a Nikon inverted Eclipse Ti microscope with built-in NIS-Elements Advanced Research software (Nikon, Japan) at the Technical Research Services of the Universitat de Girona.
DNA binding assay
A 1135-base-pair dsDNA fragment corresponding to the GFP gene fused to Apoptin at a concentration of 25 μg/ml was incubated in the presence of 300 μg/ml of each H6-Apoptin variant for 20 min in PBS. After incubation, the mixture was analyzed by agarose gel electrophoresis (1% w/v) in a TAE buffer and stained with ethidium bromide. Binding to DNA was detected by the retardation of the DNA migration in the electrophoresis.
Results
Characterization of H6-apoptin
H6-Apoptin forms water-soluble multimers that elute at the void volume when subjected to size exclusion chromatography (SEC) (Fig. 1a), as previously reported for other Apoptin variants [17, 18]. TEM images show that H6-Apoptin aggregates are spherical-like structures with radii between 10 and 12.5 nm (Fig. 1b). Characterization using DLS confirmed that H6-Apoptin exists as a single solute species with an average RH of 10.3 ± 0.16 nm, which corresponds to a molecular mass of 770 kDa and approximately 50 subunits per particle. This value is very similar to the 30–40 monomers that form the multimeric globules of Apoptin fused to MBP or to a C-terminal His-tag [4].

Purified H6-Apoptin and H6-ApopΔPro aggregate as water-soluble multimers whereas H6-ApopΔProΔLeu consists of a mixture of a monomer and a minor population of dimeric species. (a) SEC analysis of purified H6-Apoptin. H6-Apoptin elutes at the void volume of the column (arrow). (b) TEM analysis of Apoptin aggregates. Inside bar indicates the scale. (c) SEC analysis of purified H6-ApopΔPro (dotted line) and H6-ApopΔProΔLeu (continuous line). H6-ApopΔPro elutes at the void volume of the column (arrow) whereas H6-ApopΔProΔLeu elutes with forms compatible with 29 and 52 kDa. The column was calibrated with globular proteins of known molecular weight (ICN Biomedicals, Ohio, USA): cytochrome C (13 kDa), myoglobin (18 kDa), chymotrypsinogen A (24 kDa), ovalbumin (45 kDa) and BSA (67 kDa) (square regression coefficient (r2) of 0.981). (d) SDS-PAGE analysis of purified H6-ApopΔPro and of the minor (1) and major (2) fractions of H6-ApopΔProΔLeu eluted from the SEC
We compared the cytotoxic capacity of transfected and exogenously-added H6-Apoptin on NCI-H460 tumor cells. The transfected Apoptin gene was found to be cytotoxic, but when the protein was added exogenously, there was no effect (results not shown).
Production of a non-multimeric soluble form of Apoptin
We reasoned that reducing the tendency of Apoptin to aggregate might allow the protein to get into the cells, and thus provide an alternative delivery strategy which could be of considerable potential interest. With this aim, two new truncated variants were designed: the first variant, H6-ApopΔPro, lacks the PRS while the second variant, H6-ApopΔProΔLeu, lacks both the PRS and the LRS (Supplementary Fig. 1S). Like H6-Apoptin, H6-ApopΔPro behaves as a large water-soluble multimer, since it elutes in the void volume of the Superdex-75 column. The average RH of these aggregates, measured by DLS, is 9.4 ± 0.35 nm, which corresponds to an estimated molecular mass of 630 kDa and, again, to approximately 50 subunits per particle. In contrast, H6-ApopΔProΔLeu was found to be present mainly as a protein which had an elution volume of 12 ml, along with a minor fraction that eluted at 10 ml (Fig. 1c). These elution volumes correspond to molecular masses of 29 kDa for the major fraction (lower molar mass, or LMM form) and to 52 kDa for the minor one (higher molar mass, or HMM form) assuming that they were globular proteins (see below). In the SDS-PAGE (Fig. 1d), the LMM form presented a unique band of 10–11 kDa whereas the HMM form presented two bands of 10–11 kDa and 21 kDa. These results correspond to the expected molecular masses for monomeric and dimeric species. The mean RH values measured for the two forms were 2.75 ± 0.11 nm and 3.46 ± 0.27 nm; hence, if these forms were globular proteins, the estimated molecular masses would be 35.9 kDa and 62.7 kDa, respectively. However, a number of different SEC experiments have shown that these two variants correspond to a monomer and a dimer, respectively [19]. Both forms were treated with guanidinium chloride and TCEP and analyzed by SEC. The LMM form eluted as a single peak at the same elution volume (12 ml); however, a significant fraction of the HMM form converted into the LMM form.
Characterization of the biological properties of H6-ApopΔProΔLeu
It was possible that truncation had reduced or altered the biological properties exhibited by Apoptin. In order to evaluate the biological properties of the truncated variant, we focused on the major fraction, i.e., that of lower molecular mass.
First, we tested the variant’s ability to bind to DNA, since it has been postulated that this ability may be related to Apoptin’s apoptotic activity [11]. In a qualitative assay, each variant was incubated with a fragment of dsDNA. H6-Apoptin and H6-ApopΔPro bound to DNA, creating aggregates that were unable to enter the agarose gel (Supplementary Fig. 3S). DNA incubated with H6-ApopΔProΔLeu was observed as a smear band in the gel, which may indicate that this variant also bound to DNA, although to a lesser extent.
We also carried out tests to determine the subcellular localization of truncated ApopΔProΔLeu in both cancer and normal cells. To do so, the Apoptin and ApopΔProΔLeu genes, fused to GFP, were transfected into tumor cells (NCI-H460) and into normal cells (CCD-18Co). Subcellular localization was determined by confocal microscopy. GFP-Apoptin was found to be only localized in the nucleus of tumor cells while in normal cells a fraction (44%) was also visible in the cytosol (Fig. 2a, b). In contrast, GFP-ApopΔProΔLeu had a higher tendency to localize in the cytoplasm, especially in normal cells. This indicates that, although ApopΔProΔLeu’s overall tendency to accumulate in the nucleus is weakened, it retains the remarkable ability of wild-type Apoptin to selectively accumulate more readily in the nuclei of cancer cells in comparison with normal cells.

Intracellular localization of transfected GFP-Apoptin and GFP-ApopΔProΔLeu. (a) Subcellular localization of GFP-Apoptin and its truncated variant GFP-ApopΔProΔLeu in NCI-H460 tumor cells (left) and in CCD-18Co normal cells (right). Scale bar: 30 μm. (b) Subcellular distribution between the nucleus and the cytoplasm of GFP-Apoptin and GFP-ApopΔProΔLeu in NCI-H460 tumor and CCD-18Co normal cells. A total of 40 cells were analyzed to construct the figure
Next, we investigated whether ApopΔProΔLeu maintains the cytotoxic properties of wild-type Apoptin by transfecting both GFP-fused genes into NCI-H460 and 1BR.3.G cells (low levels of transfection precluded the use of CCD-18Co in this analysis). Transfected cells were selected via GFP detection using flow cytometry. From this group, dead cells were detected by PI staining. Both GFP-fused proteins killed tumor cells, although the effect of the GFP-Apoptin was 1.9 times higher than that of GFP-ApopΔProΔLeu (Fig. 3a). In contrast, neither GFP-Apoptin nor GFP-ApopΔProΔLeu produced a significant effect on normal cells (Fig. 3a).

GFP-ApopΔProΔLeu is cytotoxic for cancer cells. (a) Apoptotic activity of GFP-Apoptin (light gray bar) and GFP-ApopΔProΔLeu (black bar) transfected into NCI-H460 cancer cells and into 1BR.3.G normal cells. The percentages of dead transfected cells given are the mean values from three different experiments. Values were analyzed from 10,000 total events. (b) H6-ApopΔProΔLeu induces apoptosis specifically in tumor cells (arrows). The field is representative of n = 3 independent analyses. Scale bar: 30 μm
We transfected GFP-Apoptin and GFP-ApopΔProΔLeu into NCI-H460 and CCD-18Co cells and examined the nuclear changes associated with apoptosis using confocal microscopy after staining DNA with Hoechst 33258. In NCI-H460 transfected cells, both treatments induced nuclear morphological changes typical of apoptosis, such as chromatin condensation and nuclear fragmentation (Fig. 3b). In contrast, in CCD-18Co transfected cells, no such morphological changes were observed and cellular apoptosis was not significantly increased relative to cells transfected with GFP as a negative control. Taken as a whole, these results indicate that the truncated form of Apoptin retained the biological properties of the parental protein, even though it was somewhat less active.
H6-ApopΔProΔLeu protein is selectively cytotoxic to cancer cells when it is exogenously added
The results from the MTT cell viability assay show that H6-ApopΔProΔLeu is cytotoxic for NCI-H460 (IC50 = 7.5 μM ± 1.3), K-562 (IC50 = 13 μM ± 0.6), OVCAR-8 (IC50 = 25 μM ± 0.01) and Jurkat tumor cell lines (IC50 = 14 μM ± 0.36) (Fig. 4a). In contrast, the truncated variant does not produce any appreciable toxic effects on 1BR.3.G and CCD-18Co normal cells (Fig. 4a).

H6-ApopΔProΔLeu is cytotoxic for cancer cells when exogenously added. (a) Metabolic effect of different concentrations of H6-ApopΔProΔLeu on tumor and normal cell lines determined by the MTT assay. The tumor cell lines are OVCAR-8 (□), K-562 (■), Jurkat (▽) and NCI-H460 (▼); the normal cell lines are CCD-18Co (●) and 1BR.3.G (○). Cell growth is expressed as the percentage of control activity using the absorbance values. The curves in the figure are from one representative experiment. Equivalent results were found in at least three independent experiments. (b) Activation of procaspase-3, −8, and −9 in NCI-H460 cells treated with 15 μM of H6-ApopΔProΔLeu for 24, 36 and 48 h (dark gray). Black bars indicate untreated cells (control). Parallel samples were treated with the specific inhibitors of caspase-3, −8, and −9 (light gray) as negative controls (z-DEVD-FMK, z-IETD-FMK or z-LEHD-FMK). Results are expressed as the mean values ± SE of three independent experiments
We also investigated whether the cell death induced by treatment with H6-ApopΔProΔLeu was the result of apoptosis on NCI-H460 cells. Quantification of the percentage of NCI-H460 cells in early and late apoptosis – by FACS analysis, 24, 36 and 48 h after treatment (Table 1) – demonstrated the induction of apoptosis by the translocation of phosphatidylserine to the external hemimembrane. Apoptosis was evident at 36 h of treatment and increased further at 48 h. In addition, H6-ApopΔProΔLeu was found to induce the activation of procaspases-3, −8 and −9 (Fig. 4b), reaching a maximum level at 36 h. In the presence of caspase-specific inhibitors, this activity fell to the level of the control extracts.
Discussion
Apoptin is a very promising candidate as an antitumor drug, although its tendency to form large aggregates limits its therapeutic effectiveness. A number of strategies for delivering Apoptin into tumor cells are under development and most of them are based on the transfection of the Apoptin gene. However, these approaches have certain drawbacks: insertional mutagenesis can occur in transfected cells; the method of delivery can cause toxicity or it may lack efficacy due to transient expression. In this paper, we have presented an alternative delivery strategy that uses a soluble, non-aggregation prone variant of Apoptin to generate a cytotoxic response specifically in tumor cells.
It has been shown previously that removal of the first 65 N-terminal residues of Apoptin fused to MBP generated a variant of low apparent molecular weight which could exist in solution in equilibrium between a monomer and a dimer or a trimer [4]. This was an indication that both the proline and leucine-rich segments (PRS, residues 8 to 28 and LRS, 33 to 46) could be the determinants responsible for Apoptin’s aggregation in vivo. We have now shown that deleting residues 1 to 43, but not the smaller deletion of residues 1 to 28, eradicates Apoptin’s ability to aggregate and confirms the role of the LRS in this process.
The HMM form of H6-ApopΔProΔLeu is a monomer (10.8 kDa) although it exhibits the behavior of a protein of around 33 kDa. Nevertheless, it should be remembered that Apoptin is poorly structured [4, 19] and it is known that unfolded or partially folded proteins elute at anomalous volumes in the SEC and have higher RH in DLS [20]. Therefore, these calculations may well overestimate the size of H6-ApopΔProΔLeu species and should be considered as upper limits.
Regarding the DNA binding experiments, the lower retardation of the DNA when incubated with H6-ApopΔProΔLeu (compared to H6-Apoptin and H6-ApopΔPro) does not necessarily reflect a higher K D. It should be borne in mind that Apoptin is a multimer and this may contribute significantly to retarding DNA mobility upon binding. Moreover, binding to DNA may be increased by an avidity effect since it is known that Apoptin multimers have about eight DNA binding sites [5].
GFP-ApopΔProΔLeu has a decreased capacity to accumulate inside the nucleus compared to GFP-Apoptin, but it maintains the differential subcellular localization of the protein in tumor and normal cells. In this regard, it is interesting to note that the LRS has been implicated in the nuclear accumulation of Apoptin in both tumor and normal cells [9].
Some of the biological properties of Apoptin seem to be slightly diminished in ApopΔProΔLeu in agreement with the results described for an Apoptin variant lacking the N-terminal region (deletion of residues 1–79) [21]. However, transfection of tumor cells with the GFP-ApopΔProΔLeu gene still manages to produce up to half of the cytotoxic effect on tumor cells that is obtained with GFP-Apoptin. Furthermore, we show for the first time here that the cytotoxic effect is specific for tumor cells and, like GFP-Apoptin, this truncated variant does not affect normal cells. The results obtained for H6-ApopΔProΔLeu underline the biological importance of the central and C-terminal portions of Apoptin since large segments of the N-terminal region can be deleted with only a minor decrease in tumor selective cytotoxicity.
In vitro, exogenously added H6-ApopΔProΔLeu is cytotoxic for tumor cells (Fig. 4a) and may be an interesting candidate for evaluation as an antitumor drug. The IC50 values of H6-ApopΔProΔLeu range from 7 to 25 μM, depending on the tumor cell line assayed, which, despite being in the μM range, is a promising result for a protein drug. For example, drugs based on the ribonuclease Onconase® have reached clinical phases II and III with similar cytotoxicities against cancer cells and with much higher toxicities for non-tumor cells (for a review, see [22]). Although H6-ApopΔProΔLeu cannot yet be considered the optimal protein, it may well serve as the basis for the development of new protein drugs based on Apoptin that can, for example, easily enter into cells.
Other laboratories have described strategies to deliver the Apoptin protein into tumor cells by fusing it to a protein transduction domain (PTD) [12, 23, 24]. Comparison of the in vitro antitumor efficiencies of some of these proteins, TAT-Apoptin and PTD4-Apoptin, with H6-ApopΔProΔLeu is difficult since no cytotoxicity curves were presented. For Apoptin-hC-SOD3 construct, however, the IC50 are similar to those of H6-ApopΔProΔLeu (with an IC50 around 4 μM in HeLa cells) [23]. More recently, degradable polymeric nanocapsules [25] have been developed to deliver Apoptin into cells and IC50 values of between 30 and 100 nM have been obtained. Nevertheless, the use of such strategies in clinic may be hampered by the strong tendency of the full-length protein to aggregate. Protein aggregation may impair biological activity and also constitutes a major risk factor of formation of anti-drug antibodies [26]. The use of the truncated protein may help to overcome this drawback.
Apoptin possesses a net positive charge and its isoelectric point is estimated to be 10.3. Like other cationic proteins that internalize after being exogenously added to tumor cells (such as, for example various pancreatic ribonucleases – for a review, see [27]), it seems plausible that Apoptin is able to cross the lipid bilayer and then exert its biological action inside the cell. Our results indicate that the mechanism of cytotoxicity of H6-ApopΔProΔLeu does not require aggregation, as has been proposed for Apoptin. It has been reported that even though both N- and C-terminal regions of Apoptin have inherent cell-killing activity [6], the C-terminus is stronger at inducing apoptosis. It is likely that only the cytotoxicity that is specifically related to the N-terminal region would be dependent on protein aggregation.
In summary, we have produced and characterized a truncated form of Apoptin that retains the selective cytotoxic activity of the full protein. This variant is cytotoxic for cancer cells when exogenously added, thus providing a new means of delivering Apoptin and one that potentially overcomes the drawbacks of current gene transfection methods.
References
Prasetyo AA, Kamahora T, Kuroishi A et al (2009) Replication of chicken anemia virus (CAV) requires apoptin and is complemented by VP3 of human torque Teno virus (TTV). Virology 385:85–92. doi:10.1016/j.virol.2008.10.043
Maddika S, Mendoza FJ, Hauff K et al (2006) Cancer-selective therapy of the future: apoptin and its mechanism of action. Cancer Biol Ther 5:10–19. doi:10.4161/cbt.5.1.2400
Poon IKH, Oro C, Dias MM et al (2005) A tumor cell-specific nuclear targeting signal within chicken anemia virus VP3/apoptin. J Virol 79:1339–1341. doi:10.1128/JVI.79.2.1339-1341.2005
Leliveld SR, Zhang Y-H, Rohn JL et al (2003) Apoptin induces tumor-specific apoptosis as a globular multimer. J Biol Chem 278:9042–9051. doi:10.1074/jbc.M210803200
Leliveld SR, Dame RT, Mommaas MA et al (2003) Apoptin protein multimers form distinct higher-order nucleoprotein complexes with DNA. Nucleic Acids Res 31:4805–4813
Danen-Van Oorschot AAAM, Zhang Y-HH, Leliveld SR et al (2003) Importance of nuclear localization of apoptin for tumor-specific induction of apoptosis. J Biol Chem 278:27729–27736. doi:10.1074/jbc.M303114200
Rollano Peñaloza OM, Lewandowska M, Stetefeld J et al (2014) Apoptins: selective anticancer agents. Trends Mol Med 20:519–528. doi:10.1016/j.molmed.2014.07.003
Rohn JL (2002) A tumor-specific kinase activity regulates the viral death protein apoptin. J Biol Chem 277:50820–50827. doi:10.1074/jbc.M208557200
Poon IKH, Oro C, Dias MM et al (2005) Apoptin nuclear accumulation is modulated by a CRM1-recognized nuclear export signal that is active in normal but not in tumor cells. Cancer Res 65:7059–7064. doi:10.1158/0008-5472.CAN-05-1370
Zhou S, Zhang M, Zhang J et al (2012) Mechanisms of apoptin-induced cell death. Med Oncol 29:2985–2991. doi:10.1007/s12032-011-0119-2
Leliveld SR, Dame RT, Rohn JL et al (2004) Apoptin’s functional N- and C-termini independently bind DNA. FEBS Lett 557:155–158
Guelen L, Paterson H, Gäken J et al (2004) TAT-apoptin is efficiently delivered and induces apoptosis in cancer cells. Oncogene 23:1153–1165. doi:10.1038/sj.onc.1207224
Zhang M, Guller S, Huang Y (2007) Method to enhance transfection efficiency of cell lines and placental fibroblasts. Placenta 28:779–782. doi:10.1016/j.placenta.2007.01.012
Castro J, Ribó M, Puig T et al (2012) A cytotoxic ribonuclease reduces the expression level of P-glycoprotein in multidrug-resistant cell lines. Invest New Drugs 30:880–888. doi:10.1007/s10637-011-9636-2
Castro J, Ribó M, Navarro S et al (2011) A human ribonuclease induces apoptosis associated with p21WAF1/CIP1 induction and JNK inactivation. BMC Cancer 11:9. doi:10.1186/1471-2407-11-9
Gras SL, Waddington LJ, Goldie KN (2011) Transmission electron microscopy of amyloid fibrils. Methods Mol Biol 752:197–214. doi:10.1007/978-1-60327-223-0_13
Noteborn MH, Todd D, Verschueren CA et al (1994) A single chicken anemia virus protein induces apoptosis. J Virol 68:346–351
Zhang YH, Leliveld SR, Kooistra K et al (2003) Recombinant apoptin multimers kill tumor cells but are nontoxic and epitope-shielded in a normal-cell-specific fashion. Exp Cell Res 289:36–46. doi:10.1016/S0014-4827(03)00188-5
Ruiz-Martínez S, Pantoja-Uceda D, Castro J et al (2017) Insights into the mechanism of Apoptin’s exquisitely selective anti-tumor action from atomic level characterization of its conformation and dynamics. Arch Biochem Biophys 614:53–64. doi:10.1016/j.abb.2016.12.010
Gualfetti PJ, Iwakura M, Lee JC et al (1999) Apparent radii of the native, stable intermediates and unfolded conformers of the alpha-subunit of tryptophan synthase from E. coli, a TIM barrel protein. Biochemistry 38:13367–13378
Rohn JL, Zhang Y-H, Leliveld SR et al (2005) Relevance of apoptin’s integrity for its functional behavior. J Virol 79:1337–1338. doi:10.1128/JVI.79.2.1337-1338.2005
Pavlakis N, Vogelzang NJ (2006) Ranpirnase-an antitumour ribonuclease: its potential role in malignant mesothelioma. Expert Opin Biol Ther 6:391–399. doi:10.1517/14712598.6.4.391
Zhao J, Gao P, Xiao W et al (2011) A novel human derived cell-penetrating peptide in drug delivery. Mol Biol Rep 38:2649–2656. doi:10.1007/s11033-010-0406-6
Sun J, Yan Y, Wang X-T et al (2009) PTD4-apoptin protein therapy inhibits tumor growth in vivo. Int J Cancer 124:2973–2981. doi:10.1002/ijc.24279
Zhao M, Hu B, Gu Z et al (2013) Degradable polymeric nanocapsule for efficient intracellular delivery of a high molecular weight tumor-selective protein complex. Nano Today 8:11–20. doi:10.1016/j.nantod.2012.12.003
Ring J, Seifert J, Jesch F, Brendel W (1977) Anaphylactoid reactions due to non-immune complex serum protein aggregates. Monogr Allergy 12:27–35
Benito A, Vilanova M, Ribó M (2008) Intracellular routing of cytotoxic pancreatic-type ribonucleases. Curr Pharm Biotechnol 9:169–179
Acknowledgements
We are very grateful to Dr. Malvash Tavassoli (King’s College, UK) for providing us with the Apoptin gene, to Dr. Bruno Beaumelle (CPBS, France) for providing us with K-562 cell line and to Dr. J. L. Corchero (Autonomous University of Barcelona, Spain) for help with the DLS experiment. S. R-M gratefully acknowledges his FPU fellowship from the MEC, Spain.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
The work was supported by grants BFU2009-06935, BIO2013-43517, SAF-2013-49179-C2-2-R, QTC2014-52633-P and UNGI 10-4E-417 from MINECO (Spain) and MPCU2016/18 and SING12/0 from UdG.
Ethical approval
This article does not contain any studies performed by any of the authors involving human or animal participants.
Informed consent
For this type of study, formal consent is not required.
Electronic supplementary material
ESM 1
(DOCX 2694 kb)
Rights and permissions
About this article

Cite this article
Ruiz-Martínez, S., Castro, J., Vilanova, M. et al. A truncated apoptin protein variant selectively kills cancer cells. Invest New Drugs 35, 260–268 (2017). https://doi.org/10.1007/s10637-017-0431-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-017-0431-6