
Abstract
Apoptin, a 13.6-kD protein encoded by chicken anemia virus, is paid more and more attention, since it selectively induces apoptosis in tumor cells while abolishes cytotoxic effect in normal cells. In addition, Apoptin shows different localization in tumor cells and normal cells: it predominantly accumulates in nucleus of tumor cells, whereas in normal cells, it is detected mainly in cytoplasm. There are various mechanisms implicated in the program of Apoptin-mediated cell death. Up to now, the interpretations have been recognized including that the particular domains control nucleocytoplasmic shuttling of Apoptin, phosphorylation on specific residue and varies relevant signaling contribute to Apoptin’s activity, and the partners interacted with Apoptin regulate activity or subcellular localization of Apoptin. In this review, we make a comprehensive survey of the existing evidence about mechanisms of Apoptin’s action, which might provide scientific basis to make progress in novel targeted tumor therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chicken anemia virus (CAV) is a small virus that causes cytopathogenic effects in young chicken thymocytes via CAV-induced apoptosis. As three overlapping open reading frames are comprised, proteins VP1 (51.6 kDa), VP2 (24.0 kDa), and VP3 (13.6 kDa) are encoded in the genome [1]. Importantly, expression of VP3 alone is sufficient to induce apoptosis in a variety of cells, which is also renamed as Apoptin [1–3]. It is interesting to note that Apoptin selectively induces apoptosis in diverse cancer cells including hepatoma, osteosarcoma, melanoma, cholangiocarcinoma, colon carcinoma, lung cancer, breast cancer, prostate cancer, cervix cancer, gastric cancer, and so on [4–13], but has no effect on cell death of normal cells. To date, the mechanisms of Apoptin-induced tumor-specific cell apoptosis are not clearly understood. Based on the existing evidence reported in studies, the mechanisms of Apoptin will be summarized in detail in this report.
Nucleocytoplasmic shuttling of Apoptin in tumor and normal cells
Tumor cells are sensitive to Apoptin-induced apoptosis, whereas the corresponding normal cells are resistant. Almost all the research subscribe to an interpretation that the tumor-selective cytotoxicity of Apoptin refers to that, in tumor cells, Apoptin shows predominantly nuclear localization, but in normal cells, it is detected mainly in cytoplasm. A possible cause for Apoptin’s distinctive subcellular localization is two principal sequences, including nuclear localization signals (NLSs) and nuclear export signals (NESs).
Apoptin is a 121-amino acid protein, which shows no homology to any known cellular proteins [14]. The C-terminus of Apoptin contains a bipartite-type nuclear localization signal: NLS1 located at amino acids (aa) 82–88, and NLS2 located at aa 111–121 [15, 16]. Moreover, the residues aa 33–46 in Apoptin’s sequence is known as a putative NES [17], as shown in Fig. 1. The C-terminus of Apoptin (residues aa 74–121), out of the context of the full-length protein, represents a unique tumor cell–enhanced nuclear targeting module [16]. Danen-Van et al. used a mutagenesis strategy to explore the role of nuclear localization for Apoptin-induced cell death in tumor cells, and they found a strong correlation of nuclear localization with cell-killing activity [15]. It is worthwhile to note that they employed a construct (NLS-Apoptin) to enforce nuclear localization of Apoptin in normal human skin fibroblasts (VH10), but NLS-Apoptin was unable to induce apoptosis in VH10 cells. It implies that nuclear localization is not sufficient for Apoptin’s killing activity, and other events must be involved. Surprisingly, Wadia et al. [18] regulated the expression levels of GFP-Apoptin70−121 in either transformed 3T3ras fibroblasts or primary human foreskin fibroblasts and found an amazing switch from a predominant nuclear localization at high levels to distribution throughout the cell under low levels in both cells. Therefore, they drew the conclusion that NLS of Apoptin is not tumorigenic selective, but rather concentration dependent.

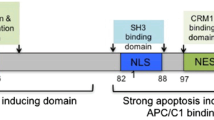
The sequence of Apoptin: NES located at amino acids 33–46 is underlined or marked in green. Two NLSs at amino acids 82–88 (NLS1) and 111–121 (NLS2) are also underlined or marked in blue
In addition, sequence analysis shows that Apoptin contains a putative CRM1-mediated NES (aa 33–46). Further investigations found that the NES is not a real NES, but in fact a cytoplasm retention signal, which is responsible for cytoplasm retention of Apoptin in normal cells, by using a specific inhibitor of the CRM1-mediated export process [17]. Then, another study uses quantitative approaches and isogenic tumor/nontumor cell pairs to conduct a comprehensive research on the most attractive domains on Apoptin. They proved that NES (aa 33–46) contributes to nuclear accumulation rather than to cytoplasmic retention or nuclear export and renamed it as the leucine-rich sequence (LRS). The actually responsible NES lies within aa 97–105 and is recognized by CRM1 [19]. Nucleocytoplasmic transport of Apoptin in tumor and normal cells is determined by a set of unique targeting signals, which comprise a bipartite-type tumor cell–specific nuclear targeting signal (NLS1 and NLS2), a CRM1-recognized NES by regulating the activity of the threonine 108 phosphorylation site (Thr-108), and an LRS by functioning as a nuclear retention sequence (as shown in Fig. 2).

The functional domains of Apoptin: LRS located on amino acids is marked in white, NLS1 at 82–88 and NLS2 at 111–121 are marked in blue, NES at 97–105 is marked in red, Thr-108 adjacent to NES is marked in yellow
Another interesting finding was proposed by Heckl [20]. These experiments used fluorescein isothiocyanate (FITC) and dansyl-labeled conjugates, which comprise the truncated C-terminal part of Apoptin (aa 81–121) with either phosphorylated or nonphosphorylated Thr-108, to study the subcellular localization of Apoptin in the tumor/normal cell pairs from the brain, prostate, and bladder. Surprisingly, all the conjugates show nuclear accumulation and cause cell death in both tumor and nontumor cells. These results contrast to a previous report, which described that enforcing nuclear uptake of Apoptin did not induce apoptosis in healthy cells (VH10) [15]. The debate may be due to differences in the composition of the cell membrane, which exists between the tumor and nontumor cell type pairs in diverse researches, and also may refer to some other unknown cell line–specific factors. And more importantly, the NLS2 is principal to nucleocytoplasmic shuttling of the FITC-labeled Apoptin81−121 peptide. It is confirmed by altering five arginines to serines in NLS2, which almost completely abolished the cellular and nuclear uptake of Apoptin81−121.
The threonine 108 phosphorylation site (Thr-108)
As we know, the above-mentioned C- and N-terminal apoptosis domains are not sufficiently responsible for the tumor-specific killing effects of Apoptin. Therefore, a further tumor-selective activation process is required. Early in 2002 [21], a speculation, that phosphorylation on residue Thr-108 appears to assist tumor-specific nuclear accumulation of Apoptin, has been proposed. In the report, by the results that the phosphorylation of Apoptin on Thr-108 was detected specifically in different original tumor and transformed cells, it has been proved that the activation of Thr-108 is relevant to a transformed or tumorigenic environment. To further assess a link between Thr-108 and Apoptin function, they constructed a phospho-mimic Apoptin mutant (mutating Thr-108 to a glutamic acid as T108E) in human diploid VH10 fibroblasts. The result showed that T108E not only translocates to the nucleus but also induces apoptosis in normal cells. Hence, it is presumed that modification on Thr-108 may act as a bifunctional switch, which includes the transportation to nucleus and the induction of apoptosis. In further in-depth studies, Lee et al. [22] and Heilman et al. [23] obtained similar results that Apoptin with Thr108 phosphorylation is not required for its tumor-specific nuclear accumulation, in contrast to the work by Poon et al. [19]. Through replacing both Thr-108 and Thr-107 with alanines to abolish the Thr108 phosphorylation site of HA-Apoptin, they examined the subcellular localization of this mutant Apoptin in HeLa cells and corresponding normal cells by immunofluorescent microscopy. It was found that the special mutant did not affect its different subcellular localization in normal and tumor cells. Moreover, abolishing the Thr-108 phosphorylation of Apoptin affected its apoptotic activity partially. One possible explanation for such consequences is that Apoptin Thr-108 phosphorylation may only be required for the apoptotic activity mediated through the C-terminal apoptosis domain. However, Apoptin exerts its apoptotic effect through both N- and C-terminal apoptosis regions.
Apoptin-mediated relevant signaling
Apoptin induce apoptosis in a P53-independent way
In spite of that mostly chemotherapy and radiotherapy have been verified to induce tumor cell apoptosis in a p53-dependent manner [24, 25], Apoptin can induce apoptosis in human tumor cells without the involvement of p53 [26]. In contrast to the tumor suppressor p53, Bcl-2, belonging to a family of Bcl-2-like proteins, acts as an apoptosis inhibiter [27]. However, the activity of Apoptin-induced apoptosis is even enhanced by overexpressing Bcl-2 [28]. A potential interpretation may be that Apoptin can activate caspases to cleave Bcl-2 into a proapoptotic form of Bcl-2 [29].Similarly, Bcl-xL, another antiapoptotic protein, cannot inhibit Apoptin-induced apoptosis in head and neck squamous cell carcinoma (HNSCC) [30]. It is concluded that the mechanism of Apoptin is independently associated with p53 and Bcl-xL and also enhanced by Bcl-2.
Apoptin-induced apoptosis relevant to caspases
Caspase-mediated signaling pathway plays an important role in the process of apoptosis. In one study, Apoptin is confirmed to not activate the upstream caspases 1 and 8 in osteosarcoma cells by using several specific inhibitors. More importantly, utilizing immunofluorescence, they found that activation of the downstream caspase 3 was essential for rapid Apoptin-induced apoptosis [31]. Research about the action of Apoptin in cholangiocarcinoma has come up with similar results. When the upstream caspases were blocked by a broad-spectrum inhibitor p35, the cell death caused by Apoptin was delayed rather than completely abolished [32]. In radioresistant SQD9 human head and neck squamous carcinoma cell lines, Apoptin induced apoptosis accompanied by the release of mitochondrial cytochrome c and subsequent activation of caspase 3 [33].
The phosphatidylinositol 3-kinase (PI3-K)/Akt pathway
The phosphatidylinositol 3-kinase (PI3-K)/Akt pathway is well known as the regulator of cell survival, proliferation, growth, transcription and translation [34]. On the contrary, Maddika et al. [35] applied mass spectrometry to analyze the proteins specifically bound to Apoptin and identified p85 (the regulatory subunit of PI3-K) and Akt (a kinase downstream of PI3-K). Further investigations verify that Apoptin induce cell death in tumor cells through the constitutive activation of PI3-K and Akt. Moreover, when Akt translocated to the nucleus together with Apoptin during Apoptin-induced cell death, nuclear-Akt acted as an apoptosis stimulator rather than a repressor. It is presumed that the PI3-K/Akt pathway acts as a pro-cell death pathway in Apoptin-induced cell death. Later, Maddika et al. make a further research on the mechanism of Apoptin’s anticancer toxicity. The most important is that they identified cyclin-dependent kinase-2(CDK2), which is modulated by Akt through p21Cip1/Waf1 [36], is crucially required for Apoptin-induced cell death by phosphorylating Apoptin. They found cells with CDK2-knockdown were markedly insensitive to Apoptin. Thus, the activation of CDK2 triggered by Akt may interpret the mechanism of Apoptin-selective toxicity in tumor cells [37].
The sphingomyelin–ceramide pathway
Liu et al. have proposed that Apoptin-mediated cell death involves the modulation of the sphingomyelin–ceramide pathway lately [38]. They detected that the accumulation of tumor suppressor ceramide was upregulated by Apoptin expression in prostate cancer cells DU145, PC-3, and LNCaP. The tumor cells showed decreased sensitivity to Apoptin-induced apoptosis, which was ascribed to the downregulation of ceramide by inhibition of acid sphingomyelinase (ASMase) or by upregulation of acid ceramidase (ACDase, which deacylates ceramide to sphingosine) [38, 39]. Ceramide is known to induce cell death by either apoptosis or necrosis in tumors and either senescence or cell cycle arrest in normal cells [40]. In addition, numerous studies have indicated that ceramide serves as a second messenger for the induction of stress-activated protein kinases to maintain a balance between cell survival and death [41, 42]. Liu et al. treated cells with Ad-GFP-Apoptin and then found that Apoptin increased ceramide levels by the activation of ASMase with a concomitant decreased sphingomyelin, which can hydrolyze into ceramide [38]. In short, activation of ASMase plays a role in Apoptin-induced cell death by generating ceramide via sphingomyelin hydrolysis. They have also reported that 10 of 15 primary prostate cancer specimens show upregulated ACDase activity compared to normal adjacent tissues. Furthermore, cotreatment with the ACDase inhibitor LCL20 significantly enhanced Apoptin-induced tumor cell–killing in vitro and vivo [39].
The mitochondrial/intrinsic pathway
There is an added research which demonstrated that Apoptin triggers apoptosis by activating the mitochondrial/intrinsic pathway, and it acted independently of the death receptor/extrinsic pathway. Furthermore, the orphan steroid receptor Nur77 transmits the Apoptin-induced death signal from the nucleus to the mitochondria in tumor cells, and using specific siRNA to inhibit Nur77 activity in MCF7 cells shows strong protection against Apoptin-induced cell death [43]. As reviewed by Marsden and Strasser, the mitochondrial/intrinsic death pathway is modulated by pro- and anti-apoptotic Bcl-2 family members [44]. Nevertheless, experimental data from Maddika et al. clearly underline the protective role of both Bcl-2 and Bcl-XL against Apoptin-triggered apoptosis. Apoptin causes the loss of mitochondrial membrane potential, resulting in the release of cytochrome c and AIF via migration of Nur77 from the nucleus to the cytoplasm [43].
The partners interacting with Apoptin during Apoptin-induced apoptosis
Leliveld et al. showed that recombinant MBP-Apoptin protein was a globular multimer when inducing apoptosis. It is possible that the multimerization of Apoptin is essential for Apoptin’s original function. There are several possible explanations such as aggregation may stabilize Apoptin, or the multimerization may facilitate certain partners (DNA, RNA, chromatin, nuclear pores, etc.) to bind Apoptin moieties [45]. In a later study, the same author reported that both Apoptin’s N- and C-terminal halves separately bound DNA, albeit with reduced affinity compared to full-length Apoptin. Since both N- and C-terminal halves can also independently induce apoptosis, Apoptin’s capacity to bind DNA is considered to be associated with its biological activity [46].
In another report [47], Apoptin interacted with death effector domain-associated factor (DEDAF), which in association with death effector domain (DED)-containing pro-apoptotic proteins involved in the regulation of transcription. Interestingly, DEDAF and Apoptin are partially (about 75%) colocalized in the nucleus of tumor cells; however, both are detected predominantly in the nucleus of apoptotic cells. In normal cells, DEDAF exhibited a predominantly nuclear localization, while Apoptin was detected preferentially in the cytoplasm. Moreover, overexpression of DEDAF induced apoptosis in tumor cell lines, but not in normal fibroblasts or mesenchymal stem cells, which is similar to the action of Apoptin.
It is well documented [48] that a major Apoptin-associated polypeptide was APC1, a subunit of the anaphase-promoting complex/cyclosome (APC/C), and it is responsible for G2/M arrest and cell death induced by Apoptin in transformed cells lacking p53. Based on the data of gel-filtration analysis, Teodoro et al. suggested that Apoptin binds to APC1, resulting in the disruption of the APC/C complex and loss of its activity. Interestingly, APC1 only interacts with the C-terminal domain of Apoptin.
Through a yeast two-hybrid screen, Huo et al. [49] identified human peptidyl-prolyl isomerase-like 3 (Ppil3) as one of the Apoptin-associated proteins, which can retain Apoptin in cytoplasm even in tumor cells via a direct protein–protein interaction. Surprisingly, the expression level of intrinsic Ppil3 contributed to the nuclear–cytoplasmic distribution of Apoptin. It is proved by the result that the upregulation of Ppil3 held Apoptin in the cytoplasm of hepatocellular carcinoma (HCC) cell lines, and knockdown of Ppil3 could resume the nuclear localization of Apoptin. Another interesting finding is that the Apoptin P109A mutant could evidently impair the function of Ppil3 and hence infer that P109 of Apoptin is required for its Ppil3-dependent cytoplasmic retention.
p85, the regulatory subunit of phosphoinositide 3-kinase (PI3-K), was identified as another Apoptin-associated protein [35, 50]. Apoptin interacts with the SH3 domain of p85 in various tumor cells independent of cell type or species, through the PKPPSK sequence (aa 81–86) of Apoptin. This specific interaction may change the conformation of p85 to sequentially active PI3-K. Importantly, downregulation of the p85 expression by siRNA or inhibitors of PI3-K severely impairs both the cytotoxic activity and nuclear localization of Apoptin [50].
Luo et al. found that Apoptin interacted with heat shock cognate protein 70 (Hsc70) via its 30- to 60-amino acid region using yeast two-hybrid and immunoprecipitation approaches [51]. Hsc70, also called Hsp73 or Hsp70-8, as a specific molecular chaperone, is a member of the heat shock protein 70 (Hsp70) family [52]. Apoptin interaction with Hsc70 induces the translocation of Hsc70 from cytoplasm to nucleus in transformed HEK293T cells. In addition, Akt phosphorylation induced by Apoptin is markedly inhibited by Hsc70 knockdown. In general, Hsc70 plays a role in Apoptin-induced phosphorylation of Akt and contributes to Apoptin-induced apoptosis in transformed cells [51].
Table 1 shows that there are also other molecules that interact with Apoptin and contribute to the nuclear localization of Apoptin or its tumor-selective killing.
Summary
The characteristic of Apoptin’s tumor-selective killing activity is not determined by a single element. Based on a comprehensive investigation into mechanisms of Apoptin-mediated cell death, it is found that it is a complicated process involving Apoptin’s nucleocytoplasmic trafficking, some apoptotic pathway, modification of conformation, interaction with other molecules or proteins, and so on. In tumor or transformed cells, the signaling and Apoptin-associated molecules are involved in Apoptin-induced apoptosis, as shown in Fig. 3. Recently, Apoptin has been used alone or combined with other treatments in tumor-bearing animal model due to its character [9, 10, 13, 57]. However, further research is warranted to better illustrate the mechanism of Apoptin-induced cell death for better clinical application in tumor therapy.

The signaling and Apoptin-associated molecules are involved in Apoptin-induced apoptosis in tumor cells. Apoptin causes the loss of mitochondrial membrane potential, resulting in the release of cytochrome c by the sphingomyelin–ceramide pathway; then, cytochrome c activates caspase-mediated signaling pathway, induces apoptosis, and regulates subunit of PI3K, resulting in the activation and nuclear translocation of Akt. In the nucleus, Apoptin can associate with different interaction partners including DEDAF (death effector domain-associated factor), Nmi (N-Myc-interacting protein), CDK2 (cyclin-dependent kinase-2), Hippi (Hip-interacting protein), Nru77 (the orphan steroid receptor), and PML (promyelocytic leukemia protein). In the cytoplasm, Apoptin can interact with FADD (Fas-associated death domain protein), APC/C (anaphase-promoting complex/cyclosome), importin β1, and Bcl10
References
Noteborn MH, Todd D, Verschueren CA, de Gauw HW, Curran WL, et al. A single chicken anemia virus protein induces apoptosis. J Virol. 1994;68(1):346–51.
Maddika S, Mendoza FJ, Hauff K, Zamzow CR, Paranjothy T, et al. Cancer-selective therapy of the future: apoptin and its mechanism of action. Cancer Biol Ther. 2006;5(1):10–9.
Noteborn MH. Chicken anemia virus induced apoptosis: underlying molecular mechanisms. Vet Microbiol. 2004;98(2):89–94.
Backendorf C, Visser AE, de Boer AG, Zimmerman R, Visser M, et al. Apoptin: therapeutic potential of an early sensor of carcinogenic transformation. Annu Rev Pharmacol Toxicol. 2008;48:143–69.
Li X, Jin N, Mi Z, Lian H, Sun L, et al. Antitumor effects of a recombinant fowlpox virus expressing Apoptin in vivo and in vitro. Int J Cancer. 2006;119(12):2948–57.
Lian H, Jin N, Li X, Mi Z, Zhang J, et al. Induction of an effective anti-tumor immune response and tumor regression by combined administration of IL-18 and Apoptin. Cancer Immunol Immunother. 2007;56(2):181–92.
Tavassoli M, Guelen L, Luxon BA, Gäken J. Apoptin: specific killer of tumor cells? Apoptosis. 2005;10(4):717–24.
Olijslagers SJ, Zhang YH, Backendorf C, Noteborn MH. Additive cytotoxic effect of Apoptin and chemotherapeutic agents paclitaxel and etoposide on human tumour cells. Basic Clin Pharmacol Toxicol. 2007;100(2):127–31.
Pan Y, Fang L, Fan H, Luo R, Zhao Q, et al. Antitumor effects of a recombinant pseudotype baculovirus expressing Apoptin in vitro and in vivo. Int J Cancer. 2010;126(11):2741–51.
Jin JL, Gong J, Yin TJ, Lu YJ, Xia JJ, et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur J Pharmacol. 2011;654(1):17–25.
Olijslagers S, Dege AY, Dinsart C, Voorhoeve M, Rommelaere J, et al. Potentiation of a recombinant oncolytic parvovirus by expression of Apoptin. Cancer Gene Ther. 2001;8(12):958–65.
Yu CM, Xu HT, Du J. Recombinant Apoptin gene retrovirus induces apoptosis in human breast cancer cells 435. Chin J Cancer Res. 2009;21(3):194–201.
Li X, Liu Y, Wen Z, Li C, Lu H, et al. Potent anti-tumor effects of a dual specific oncolytic adenovirus expressing Apoptin in vitro and in vivo. Mol Cancer. 2010;9:10.
Noteborn MH, de Boer GF, van Roozelaar DJ, Karreman C, Kranenburg O, et al. Characterization of cloned chicken anemia virus DNA that contains all elements for the infectious replication cycle. J Virol. 1991;65(6):3131–9.
Danen-Van Oorschot AA, Zhang YH, Leliveld SR, Rohn JL, Seelen MC, et al. Importance of nuclear localization of apoptin for tumor-specific induction of apoptosis. J Biol Chem. 2003;278(30):27729–36.
Kuusisto HV, Wagstaff KM, Alvisi G, Jans DA. The C-terminus of Apoptin represents a unique tumor cell-enhanced nuclear targeting module. Int J Cancer. 2008;123(12):2965–9.
Wang QM, Fan GC, Chen JZ, Chen HP, He FC. A putative NES mediates cytoplasmic localization of Apoptin in normal cells. Acta Biochim Biophys Sin (Shanghai). 2004;36(12):817–23.
Wadia JS, Wagner MV, Ezhevsky SA, Dowdy SF. Apoptin/VP3 contains a concentration-dependent nuclear localization signal (NLS), not a tumorigenic selective NLS. J Virol. 2004;78(11):6077–8.
Poon IK, Oro C, Dias MM, Zhang J, Jans DA. Apoptin nuclear accumulation is modulated by a CRM1-recognized nuclear export signal that is active in normal but not in tumor cells. Cancer Res. 2005;65(16):7059–64.
Heckl S, Regenbogen M, Sturzu A, Gharabaghi A, Feil G, et al. Value of apoptin’s 40-amino-acid C-terminal fragment for the differentiation between human tumor and non-tumor cells. Apoptosis. 2008;13(4):495–508.
Rohn JL, Zhang YH, Aalbers RI, Otto N, Den Hertog J, et al. A tumor-specific kinase activity regulates the viral death protein Apoptin. J Biol Chem. 2002;277(52):50820–7.
Lee YH, Cheng CM, Chang YF, Wang TY, Yuo CY. Apoptin T108 phosphorylation is not required for its tumor-specific nuclear localization but partially affects its apoptotic activity. Biochem Biophys Res Commun. 2007;354(2):391–5.
Heilman DW, Teodoro JG, Green MR. Apoptin nucleocytoplasmic shuttling is required for cell type-specific localization, apoptosis, and recruitment of the anaphase-promoting complex/cyclosome to PML bodies. J Virol. 2006;80(15):7535–45.
Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74(6):957–67.
Warters RL. Radiation-induced apoptosis in a murine T-cell hybridoma. Cancer Res. 1992;52(4):883–90.
Zhuang SM, Shvarts A, van Ormondt H, Jochemsen AG, van der Eb AJ, et al. Apoptin, a protein derived from chicken anemia virus, induces p53-independent apoptosis in human osteosarcoma cells. Cancer Res. 1995;55(3):486–9.
Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387(6635):773–6.
Danen-Van Oorschot AA, Zhang Y, Erkeland SJ, Fischer DF, van der Eb AJ, et al. The effect of Bcl-2 on Apoptin in ‘normal’ vs transformed human cells. Leukemia. 1999;13(Suppl 1):S75–7.
Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278(5345):1966–8.
Schoop RA, Kooistra K, Baatenburg De Jong RJ, Noteborn MH. Bcl-xL inhibits p53- but not Apoptin-induced apoptosis in head and neck squamous cell carcinoma cell line. Int J Cancer. 2004;109(1):38–42.
Danen-van Oorschot AA, van Der Eb AJ, Noteborn MH. The chicken anemia virus-derived protein Apoptin requires activation of caspases for induction of apoptosis in human tumor cells. J Virol. 2000;74(15):7072–8.
Pietersen AM, Rutjes SA, van Tongeren J, Vogels R, Wesseling JG, et al. The tumor-selective viral protein Apoptin effectively kills human biliary tract cancer cells. J Mol Med (Berl). 2004;82(1):56–63.
Schoop RA, Verdegaal EM, Baatenburg de Jong RJ, Noteborn MH. Apoptin enhances radiation-induced cell death in poorly responding head and neck squamous cell carcinoma cells. Basic Clin Pharmacol Toxicol. 2010;106(2):130–4.
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–7.
Maddika S, Bay GH, Kroczak TJ, Ande SR, Maddika S, et al. Akt is transferred to the nucleus of cells treated with Apoptin, and it participates in Apoptin-induced cell death. Cell Prolif. 2007;40(6):835–48.
Jänicke RU, Sohn D, Essmann F, Schulze-Osthoff K. The multiple battles fought by anti-apoptotic p21. Cell Cycle. 2007;6(4):407–13.
Maddika S, Panigrahi S, Wiechec E, Wesselborg S, Fischer U, et al. Unscheduled Akt-triggered activation of cyclin-dependent kinase 2 as a key effector mechanism of Apoptin’s anticancer toxicity. Mol Cell Biol. 2009;29(5):1235–48.
Liu X, Zeidan YH, Elojeimy S, Holman DH, El-Zawahry AM, et al. Involvement of sphingolipids in Apoptin-induced cell killing. Mol Ther. 2006;14(5):627–36.
Liu X, Elojeimy S, El-Zawahry AM, Holman DH, Bielawska A, et al. Modulation of ceramide metabolism enhances viral protein Apoptin’s cytotoxicity in prostate cancer. Mol Ther. 2006;14(5):637–46.
Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4(8):604–16.
Charruyer A, Grazide S, Bezombes C, Müller S, Laurent G, et al. UV-C light induces raft-associated acid sphingomyelinase and JNK activation and translocation independently on a nuclear signal. J Biol Chem. 2005;280(19):19196–204.
Loidl A, Claus R, Ingolic E, Deigner HP, Hermetter A. Role of ceramide in activation of stress-associated MAP kinases by minimally modified LDL in vascular smooth muscle cells. Biochim Biophys Acta. 2004;1690(2):150–8.
Maddika S, Booy EP, Johar D, Gibson SB, Ghavami S, et al. Cancer-specific toxicity of Apoptin is independent of death receptors but involves the loss of mitochondrial membrane potential and the release of mitochondrial cell-death mediators by a Nur77-dependent pathway. J Cell Sci. 2005;118(Pt 19):4485–93.
Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105.
Leliveld SR, Zhang YH, Rohn JL, Noteborn MH, Abrahams JP. Apoptin induces tumor-specific apoptosis as a globular multimer. J Biol Chem. 2003;278(11):9042–51.
Leliveld SR, Dame RT, Rohn JL, Noteborn MH, Abrahams JP. Apoptin’s functional N- and C-termini independently bind DNA. FEBS Lett. 2004;557(1–3):155–8.
Danen-van Oorschot AA, Voskamp P, Seelen MC, van Miltenburg MH, Bolk MW, et al. Human death effector domain-associated factor interacts with the viral apoptosis agonist Apoptin and exerts tumor-preferential cell killing. Cell Death Differ. 2004;11(5):564–73.
Teodoro JG, Heilman DW, Parker AE, Green MR. The viral protein Apoptin associates with the anaphase-promoting complex to induce G2/M arrest and apoptosis in the absence of p53. Genes Dev. 2004;18(16):1952–7.
Huo DH, Yi LN, Yang J. Interaction with Ppil3 leads to the cytoplasmic localization of Apoptin in tumor cells. Biochem Biophys Res Commun. 2008;372(1):14–8.
Maddika S, Wiechec E, Ande SR, Poon IK, Fischer U, et al. Interaction with PI3-kinase contributes to the cytotoxic activity of Apoptin. Oncogene. 2008;27(21):3060–5.
Chen K, Luo Z, Tang J, Zheng SJ. A critical role of heat shock cognate protein 70 in Apoptin-induced phosphorylation of Akt. Biochem Biophys Res Commun. 2011;409(2):200–4.
Daugaard M, Rohde M, Jäättelä M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581(19):3702–10.
Sun GJ, Tong X, Dong Y, Mei ZZ, Sun ZX. Identification of a protein interacting with Apoptin from human leucocyte cDNA library by using yeast two-hybrid screening. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai). 2002;34(3):369–72.
Cheng CM, Huang SP, Chang YF, Chung WY, Yuo CY. The viral death protein Apoptin interacts with Hippi, the protein interactor of Huntingtin-interacting protein 1. Biochem Biophys Res Commun. 2003;305(2):359–64.
Guelen L, Paterson H, Gäken J, Meyers M, Farzaneh F, et al. TAT-apoptin is efficiently delivered and induces apoptosis in cancer cells. Oncogene. 2004;23(5):1153–65.
Zhong S, Salomoni P, Ronchetti S, Guo A, Ruggero D, et al. Promyelocytic leukemia protein (PML) and Daxx participate in a novel nuclear pathway for apoptosis. J Exp Med. 2000;191(4):631–40.
Sun J, Yan Y, Wang XT, Liu XW, Peng DJ, et al. PTD4-apoptin protein therapy inhibits tumor growth in vivo. Int J Cancer. 2009;124(12):2973–81.
Jiang J, Cole D, Westwood N, Macpherson L, Farzaneh F, et al. Crucial roles for protein kinase C isoforms in tumor-specific killing by Apoptin. Cancer Res. 2010;70(18):7242–52.
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 81072053) and Science and Technology Planning Project of Shaanxi Province (2010K14-02), China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhou, S., Zhang, M., Zhang, J. et al. Mechanisms of Apoptin-induced cell death. Med Oncol 29, 2985–2991 (2012). https://doi.org/10.1007/s12032-011-0119-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12032-011-0119-2