
Summary
Background MK-5108 is a potent/highly selective Aurora A kinase inhibitor. Methods A randomized Phase I study of MK-5108, administered p.o. BID Q12h on days 1–2 in 14–21 day cycles either alone (MT; Panel1/n = 18; 200 to 1800 mg) or in combination (CT; Panel2/n = 17; 100 to 225 mg) with IV docetaxel 60 mg/m2, determined the maximum tolerated dose (MTD), pharmacokinetics (PK), pharmacodynamics (Panel1, only) and tumor response in patients with advanced solid tumors. This study was terminated early due to toxicities in Panel2 at MK-5108 doses below the anticipated PK exposure target. Results 35 patients enrolled (33 evaluable for tumor response). No dose-limiting toxicities (DLTs) were observed in Panel1; three patients had 3 DLTs in Panel2 (G3 and G4 febrile neutropenia at 200 and 450 mg/day, respectively; G3 infection at 450 mg/day). In Panel1, AUC0-12hr and Cmax increased less than dose proportionally following the first MT dose but increased roughly dose proportionally across 200 to 3600 mg/day after 4th dose. The t1/2 ranged from 6.6 to 13.5 h across both panels. No clear effects on immunohistochemistry markers were observed; however, significant dose-related increases in gene expression were seen pre-/post-treatment. Best responses were 9/17 stable disease (SD) (Panel1) as well as 1/16 PR and 7/16 SD (Panel2) (450 mg/day). Conclusions MK-5108 MT was well tolerated at doses up to 3600 mg/day with plasma levels exceeding the minimum daily exposure target (83 μM*hr). The MTD for MK-5108 + docetaxel (CT) was established at 300 mg/day, below the exposure target. Use of pharmacodynamic gene expression assays to determine target engagement was validated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aurora A kinase (AK-A) is a serine/threonine kinase required for entry into and progression through mitosis. During the mitotic cycle, Aurora A regulates the initiation of mitosis in addition to playing a critical role in centrosome maturation, establishment of bipolar spindles during cell division and chromosome attachment to the spindle [1]. The expression of Aurora A is tightly regulated throughout the cell cycle, peaking during late G2 and mitosis whereas resting cells have low or undetectable levels of this enzyme. Dysfunctional regulation of this protein may lead to genetic instability, thereby potentially contributing to tumorigenesis. AK-A is considered an oncogene and the human AURKA gene that encodes AK-A is located in chromosome 20q13, a known hotspot for gene amplification in various cancer types. Several studies have demonstrated amplification of the AURKA gene locus and overexpression of AK-A in various tumors [2–4]. Therefore, inhibition of AK-A represents a rationale target for anticancer therapy and several such agents are currently being evaluated in ongoing clinical trials [5].
Given their known mechanism of action, Aurora kinase inhibitors (AKIs) may potentiate antitumor effects when combined with various anticancer treatment modalities including both chemotherapeutic agents and radiation therapy. Several preclinical studies have shown that suppression of AK-A through the administration of chemical inhibitors or antisense RNA greatly enhances, or even restores the chemosensitivity of cancer cell lines to taxanes [6–8]. Thus the combination of an AKI plus a taxane is a logical therapeutic regimen currently under evaluation in ongoing phase I/II clinical trials.
MK-5108 is a potent adenosine triphosphate–competitive inhibitor of AK-A (IC50 0.064 nM) with a comparatively high target specificity relative to other kinases, including both Aurora B and Aurora C. In preclinical studies, MK-5108 was shown to possess modest single-agent activity in a subset of human tumor cell lines and xenograft models [9]. However, the preclinical antitumor efficacy of MK-5108 was significantly enhanced when combined with docetaxel as well as with various other antimicrotubule agents (other taxanes, vinca alkaloids, epothilones), targeted agents and chemotherapeutics [9]. Based on preclinical findings, the initial clinical development program for MK-5108 focused on its ability to sensitize tumor tissues to docetaxel.
The primary objective of this Phase I study was to investigate the safety/toxicity profile of MK-5108, administered orally both as monotherapy (MT) and in combination with docetaxel (CT), in patients with advanced and/or refractory solid tumors. Specifically, this study sought to define the dose-limiting toxicities (DLTs) and the maximum tolerated dose (MTD) of MK-5108 MT and CT. Preclinical efficacy studies indicated that AK-A inhibition for 48 h resulted in maximal sensitization to docetaxel, therefore oral doses of MK-5108 were administered every 12 h (Q12hr) for 2 days within each treatment cycle, where cycle length was 14 days for MT and 21 days for the CT regimen. Additional objectives included an investigation of the plasma pharmacokinetics (PK) of MK-5108 following oral dosing in human patients. Further, this study explored the relationships between PK parameters and specific pharmacodynamic (PD) biomarkers of AK-A inhibition in surrogate (scalp and skin hair follicle) tissues. Finally, the antitumor efficacy of MK-5108 MT and CT were assessed via imaging studies as a secondary objective in this study.
Materials and methods
Patient selection
All study participants provided written informed consent before the initiation of any study procedures. The study protocol was approved by each participating institution’s Institutional Review Board. The study was conducted in accordance with the Principles of Good Clinical Practice guidelines, the Declaration of Helsinki and other statues and regulations for the protection of the rights and welfare of people participating in biomedical research.
Eliglibe patients included individuals with histologically-confirmed metastatic or locally advanced solid tumors who failed to respond to standard therapy, progressed despite standard therapy, or had tumors for which standard therapy did not exist. Patients who received prior therapy with docetaxel were allowed to participate in this study. Other key eligibility criteria included: age ≥18 years; Eastern Cooperative Oncology Group performance status ≤2; and adequate hematopoietic (absolute neutrophil count ≥1500/μL, platelets ≥100,000/μL, hemoglobin ≥9 g/dL), hepatic (total bilirubin ≤1.5 X upper limit of normal [ULN], aspartate aminotransferase and alanine aminotransferase ≤2.5 X ULN, alkaline phosphatase ≤2.5 X ULN), renal (serum creatinine ≤1.5 X ULN or calculated creatinine clearance ≥60 mL) and coagulation (prothrombin time and partial thromboplastin time ≤1.2 X ULN) function. Exclusion criteria included: anticancer therapy within 4 weeks (6 weeks for nitrosoureas or mitomycin C) prior to entering the study; history of high-dose chemotherapy with peripheral blood or bone marrow stem cell support; treatment with ≥3 regimens of chemotherapy (≥2 if a prior regimen included carboplatin, nitrosourea, mitomycin, or gemcitabine); previous history of radiotherapy to the pelvis or more than 15 % of the bone; a primary central nervous system (CNS) tumor or active CNS metastases; known sensitvity to components of MK-5108 or docetaxel; use of pharmaceuticals known to be strong CYP3A4 inhibitors or inducers; pregnant or breastfeeding; known infections with human immunodeficiency virus, hepatitis B or C; symptomatic ascites or pleural effusions.
Study design
This was a multi-center (6 sites in the United States), randomized, open-label, dose escalation, 2-panel, Phase I study conducted between the dates of April 2008 to April 2011 in patients with advanced and/or refractory solid tumors (Merck & Co., Inc., Kenilworth, New Jersey: Protocol number MK-5108 PN001-04; clinicaltrials.gov: NCT00543387). The study was terminated to new enrollment by the sponsor in December of 2009 due to observed toxicities in the CT arm at low MK-5108 doses thus preventing achievement of the targeted therapeutic exposure range of MK-5108 when used in combination with a standard dose of docetaxel. MK-5108 was administered orally with 5 mL water as dry-filled capsules (i.e., 25, 100 or 200 mg caplets to achieve the intended dose [Merck & Co., Inc., Kenilworth, New Jersey]) following a 2-h fast. Docetaxel (TAOTERE™, SANOFI-AVENTIS US) was administered as 1-h intravenous (IV) infusion through a peripherally inserted central catheter or other centrally placed catheter at a fixed dose of 60 mg/m2 Q21 days 2 h prior to dosing with MK-5108. The fixed dose of docetaxel of 60 mg/m2 Q21 days was selected to provide a safety window for accommodating escalation of MK-5108 doses.
Patients with demonstrated disease progression or stable disease while taking MK-5108 MT were allowed to crossover to CT at the discretion of the study investigator. A total of 4 patients (22.2 %) who were initially randomized to MT crossed over to CT during this study. In both panels, MK-5108 was administered to cohorts of patients (n = 3 up to a maximum of 6) in sequentially rising doses. A total of 6 daily dose levels, administered BID Q12hr, were planned for Panel 1 (i.e., 400, 800, 1600, 2400, 3000 and 3600 mg/day. A total of eight daily dose levels, administered BID Q12hr, were planned for Panel 2 (i.e., 200, 300, 450, 650, 950, 1300, 1800 and 2400 mg/day). Panel 1 enrollment was initiated prior to Panel 2. Patients were enrolled in Panel 2 after at least one dose cohort of MK-5108 MT in Panel 1 was determined to be safe and well tolerated as defined by the absence of a Grade 2 or higher adverse event (AE) deemed to be related (definitely, probably, or possibly) to study medication by the investigator during the first treatment cycle.
Once Panel 2 was open for enrollment, new patients were randomized into a cohort in either Panel 1 or Panel 2. Dosing in the 2 panels progressed independently and concurrently in sequentially rising doses. Doses of MK-5108 in Panel 2 could not exceed those shown to be safe and well tolerated in Panel 1. Prophylatic use of granulocyte stimulating factors (e.g., pegfilgastrim or filgastrim) to counteract known hematologic AEs of docetaxel (i.e., neutropenia, leucopenia, thrombocytopenia and anemia) was not permitted in this study [10]. These agents were only permitted in patients having a neutropenic-related DLT after receiving docetaxel.
Cycle length was initially 14 days in Panel 1 but increased to 21 days if Grade 2 or higher drug-related toxicity was observed prior to or on day 15. Cycle length was 21 days in Panel 2. Of the 18 patients enrolled in Panel 1, 11 required extension of cycle length from 14 to ≥21 days. A standard 3 + 3 dose escalation scheme was utilized in both panels. The first three to six patients on the study were enrolled in the first dose-level of Panel 1. Subsequent patients were randomized to Panel 1 or 2.
Patients could be discontinued from the study for any of the following reasons: disease progression; concomitant illness preventing further administration of treatment; AEs; pregnancy; patient withdraws consent; change in therapy at investigator’s discretion; lost to follow-up.
Safety and tolerability assessments
Patients were monitored for the development of AEs, DLT, and disease progression throughout the course of the study. AEs were evaluated according to criteria outlined in the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 3.0. Determinations of dose-limiting hematologic and non-hematologic toxicities were based on events occurring during the first cycle of study drug administration. In order to be declared a DLT, an AE had to be deemed related (definitely, probably, or possibly) to study therapy by the study investigator. Hematologic dose-limiting toxicities included: any Grade 5 hematologic toxicity; Grade 4 neutropenia lasting for ≥7 days in duration; Grade 3 or Grade 4 neutropenia with fever and/or infection requiring antibiotic or anti-fungal treatment; Grade 4 thrombocytopenia. Non-hematologic dose-limiting toxicities included: any Grade 3, 4, or 5 non-hematologic toxicity in the setting of adequate supportive care, except for alopecia and hypersensitivity reactions; or any drug-related AE, regardless of CTCAE grade, leading to a dose modification of MK-5108 or a dose delay of 3 weeks.
Patients who experienced a first cycle DLT attributable to MK-5108 had their dose of MK-5108 reduced by 1 dose level for the remainder of the study, or by 50 % if currently at dose level 1. Treatment for each new cycle was delayed until drug-related toxicities resolved to Grade 0 or 1 or to the patient’s baseline level. Patients with unresolved drug-related toxicities lasting ≥3 weeks from the date of the next scheduled treatment were not permitted to continue in the study. Patients were allowed a maximum of 2 dose reductions for DLT beyond which they were discontinued from the study. Patients were not allowed to have their dose of docetaxel de-escalated below 60 mg/m2 and were discontinued from the study if dose reduction was warranted. The MTD of MK-5108 was defined as the dose level immediately below the dose level at which ≥2 patients experienced ≥1 DLT each.
Pharmacokinetics
In both Panels, serum concentrations of MK-5108 (and its potential metabolites) were measured at the following sampling points within Cycle 1, only: 1) Day 1 at predose (0; i.e., before administration of the 1st dose), and 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after the first dose of MK-5108; 2) Day 2 at predose (0; i.e., before administration of the 4th dose), and 0.5, 1, 2, 3, 4, 6, 8, 12, 20, 36, and 44 h after the fourth dose of MK-5108. The 20- and 44-h collection time points allowed a ±2 h window.
Non-compartmental pharmacokinetic parameters including area under the plasma concentration–time curves (AUC) from time 0 to the last measurable time (AUC0–t), maximum concentration (Cmax), time to maximum concentration (Tmax), and elimination half-life (t1/2) were calculated for MK-5108. Pharmacokinetic parameters were calculated using WinNonlin® software (Version 5.0.1; Pharsight Corporation, Mountain View, California, USA). The AUC0–t was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. Cmax and Tmax were obtained by visual inspection of the individual plasma concentration-time profile. The apparent terminal elimination rate constant (k) was estimated by regression of the terminal log-linear portion of the blood concentration–time profile (using quantifiable concentrations only). The apparent terminal t1/2 was calculated as the quotient of ln(2) and k.
Pharmacodynamics
Hair follicles obtained from at least 15 hair shafts and skin biopsies were evaluated for MK-5108-induced PD effects. In Panels 1 and 2, hair shafts and skin biopsies were obtained pre-dose and post-dose on Day 2 of Cycle 1 at 4 (±1) hours after the third dose of MK-5108. Hair follicles and skin biopsies were analyzed for phospho-Aurora B (pAurB) and phospho-histone H3 (pHH3) using immunohistochemistry (IHC). In addition, a panel of seven gene expression PD biomarkers specific for Aurora A inhibition (AURKA, AURKB, BIRC5, PRC1, TACC3, DLGAP5 [DLG7], and NDC80 [KNTC2]) was evaluated using the same tissue specimens by quantitative reverse transcription polymerase chain reaction methods performed on extracted hair follicle RNA. The log ratios (post-dose to pre-dose) in gene expression for these seven genes were expected to increase in response to MK-5108 treatment.
Tumor response
Tumor response was assessed during the study by radiographic (computed tomography or magnetic resonance imaging [MRI]) evaluation or physical exam. Standard serum tumor markers where appropriate for specific tumor subtypes, were obtained. Overall tumor response was assessed using Response Evaluation Criteria in Solid Tumors (RECIST) criteria version 1.0 at the designated time points. Although RECIST criteria defines progression as on a 20 % increase in the sum of the longest diameter of all target lesions; however, an increase of no more than 50 % could be used, at the investigator’s discretion, to define progressive disease for patients in this study.
A baseline evaluation was performed as close as possible to the beginning of treatment and not more than 28 days prior to the beginning of treatment. Patients were assessed for tumor responsiveness every other treatment cycle, within 1 week of the next cycle of therapy starting after Cycle 2 (unless progressive disease was documented earlier). After 3 post-treatment assessments, radiologic assessment of response was performed every third cycle (within 1 week of the next), or more frequently at the discretion of the study investigator. The same method of assessment and the same technique was employed throughout the study to characterize each of the identified and reported lesions at baseline and during follow-up. Attempts were made for the computed tomography and MRI to be performed on cuts of 5 mm or less in slice thickness contiguously. Bone scans were performed if there was known prior bony metastatic disease or a strong clinical suspicion of bony metastatic disease.
Statistical analysis
The study population for safety analyses included all patients who received at least one dose of study medication. AEs were summarized by tabulating the number (%) of patients experiencing at least one AE within each body system and within each preferred term and were analyzed based on the dose the patient was taking at the time of the AE. Numbers and percentages also were summarized by severity of the AE (mild, moderate, severe) and the potential relationship to study drug.
The DLT-evaluable population included all patients who met DLT assessment criteria. The MTD of MK-5108 was determined based on DLT occurring in the first cycle of treatment with MK-5108 MT or CT. Patients in Panel 1 were considered evaluable for the MTD if they either experienced a DLT within the first cycle, or if the patient had 14 to 21 days of follow-up after study drug administration without experiencing a DLT. Patients in Panel 2 were considered evaluable for the MTD if they either experienced a DLT within the first cycle, or if the patient had 21 days of follow-up after study drug administration without experiencing a DLT. The minimal evaluation period for dose escalation decisions was 16 to 23 days for MK-5108 MT and 23 days for MK-5108 CT. Patients were only evaluable for MTD based on the dose cohort they were randomized to at study start; therefore, patients who crossed over from Panel 1 to Panel 2 were only considered in the MTD assessment for MK-5108 MT.
The efficacy analyses as well as the PK/PD analyses were based on the population of patients who received at least 90 % of intended drug volume and had efficacy and/or PK/PD measurements at baseline and at least once during treatment. Further, the PK-evaluable population included all patients for whom PK sampling was completed on at least 1 day. Descriptive statistics were used to summarize patient characteristics, treatment administration/compliance, safety, PK parameters, and efficacy.
All patients with on-treatment imaging studies were assessed for response to treatment. Each patient was assigned to one of the following categories: 1) complete response; 2) partial response; 3) stable disease; 4) progressive disease; 5) early death from malignant disease; 6) early death from toxicity; 7) early death because of other cause; or 8) unknown (not assessable, insufficient data). All patients who met the study eligibility criteria were included in the main analysis of response rate. Patients assigned a response category of 4–8 were considered failing to respond to treatment (i.e., disease progression). The number (%) of patients in each category and the associated 95 % confidence intervals (CI) were calculated and reported.
Results
Patient demographics and accounting
Between April of 2008 and December of 2009, 35 patients with advanced and/or refractory solid tumors were enrolled and received treatment in this study. A total of 18 patients were initially enrolled into Panel 1 and received MK-5108 MT at the following dose levels: 200, 400, 800, 1200, 1500, and 1800 mg Q12hr on Days 1 and 2 of each 21-day cycle. A total of 17 patients were enrolled into Panel 2 and received MK-5108 CT with 60 mg/m2 docetaxel at the following MK-5108 dose levels: 100, 150, and 225 mg Q12hr on Days 1 and 2 of each 21-day cycle. Of the patients initially enrolled and treated with MK −5108 MT in Panel 1, 4 patients were allowed to crossover to Panel 2 and received MK-5108 CT with docetaxel. Patient demographics and baseline characteristics are summarized in Table 1. The randomized population primarily consisted of Caucasian males with Grade IV cancer and a mean age of 59 years.
The majority of patients discontinued from this study due to progressive disease (28/35; 80 %), including 12 (12/14; 85.7 %) in Panel 1 and 13 (13/17; 76.5 %) in Panel 2. One patient in Panel 1 who discontinued from the study due to progressive disesase died during the study. Of the 4 patients who crossed over from Panel 1 to 2 during the study, 3 (3/4; 75 %) discontinued early due to progressive disease. In total, 3 patients (3/35; 8.6 %) discontinued from the study due to an AE (i.e., non-serious AE of arthralgia [Panel 1: non-serious, Grade 2, not drug-related], myocardial infarction [Panel 2: serious, Grade 3, not drug-related], and angioedema [Panel 2: serious, Grade 3 AE, drug-related]). Other reasons for discontinuation included physician’s decision (2/35; 5.7 %), study termination (1/35; 2.9 %) and patient withdrawal (1/35; 2.9 %).
Safety/toxicity
All 35 treated patients experienced at least one AE during the study (Tables 2 and 3). A total of 26 (26/35; 74.3 %) patients experienced one or more drug-related AEs. The majority of these patients (24/26; 92.3 %) reported one or more Grade 3 or Grade 4 AEs during the study. A total of four patients, two patients each from Panel 1 (i.e., arthralgia and malignant neoplasm progression; 2/14 or 14.3 %) and Panel 2 (i.e., angioedema and myocardial infarction; 2/17 or 11.8 %), discontinued treatment (or had drug withdrawn) due to AEs. Only one patient in this study discontinued due to a drug-related AE (i.e., angioedema in Panel 2). One patient in the MK-5108 MT group was hospitalized due to a Grade 3, serious AE of disease progression (i.e., malignant neoplasm progression) and subsequently died during the safety follow-up period. This AE was deemed by the study investigator to be unrelated to study treatment. No patients in this study died due to an AE. Additionally, no patients had Grade 5 AEs in this study.
MK-5108 MT was generally well tolerated when administered at doses up to 1800 mg Q12hr (3600 mg/day). In Panel 1, the most common AEs observed across all grades and doses were those belonging to the system organ classes (SOCs) of Gastrointestinal disorders (12/14; 85.7 %) and General disorders and aministration site conditions (11/14; 78.6 %). Overall, combined treatment with docetaxel and MK-5108 doses up to 225 mg Q12hr (450 mg/day) was less well tolerated than MK-5108 MT. In Panel 2, the most frequent AEs across all grades and doses were those belonging to the SOCs of Blood and lymphatic system disorders (15/17; 88.2 %), General disorders and administration site conditions (15/17; 88.2 %), Gastrointestinal disorders (12/17; 70.6 %), Metabolism and nutrition disorders (12/17; 70.6 %), and Skin and subcutaneous tissue disorders (12/17; 70.6 %). These observed toxicities are consistent with the known safety and tolerability profile of docetaxel treatment. In general, the overall profile of AEs reported by the 4 patients who crossed over from Panel 1 to Panel 2 was similar to that seen in patients receiving MK-5108 CT.
The reported drug-related toxicities across all grades and doses are summarized in Table 4. The most frequent drug-related AEs observed in Panel 1 were those belonging to Gastrointestinal disorders (7/14; 50 %), General disorders and aministration site conditions (7/14; 50 %) and Blood and lymphatic system disorders (4/14; 28.6 %). No patients in Panel 1 experienced a Grade 3 or 4 drug-related AE. In Panel 2, the most frequent drug-related AEs were those belonging to the SOCs of Blood and lymphatic system disorders (14/17; 82.4 %), General disorders and administration site conditions (11/17; 64.7 %) and Gastrointestinal disorders (11/17; 64.7 %). A total of 12 (12/17; 70.6 %) patients in Panel 2 experienced a Grade 3 or 4 drug-related AE. In general, the overall profile of drug-related AEs reported by the 4 patients who crossed over from Panel 1 to Panel 2 was similar to that seen in patients receiving MK-5108 CT. Two patients in the cross over group reported Grade 3 or 4 drug-related AEs.
Determination of MTD
Summary statistics on the duration of MK-5108 therapy for patients in Panels 1 and 2 are summarized in Table 2. The median duration of MK-5108 treatment was 4.5 cycles (range: 1–35 cycles) in Panel 1 compared with 2 cycles (range: 1–13 cycles) in Panel 2.
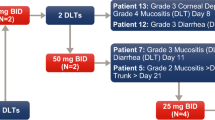
An MTD was not established for MK-5108 MT because no patients in Panel 1 experienced a DLT. In Panel 2, one patient receiving MK-5108 100 mg Q12hr (200 mg/day) CT experienced a DLT of febrile neutropenia (Grade 3). Two patients in Panel 2 experienced one DLT each of febrile neutropenia (Grade 4) and infection (Grade 3) at the 225 mg Q12hr (450 mg/day) dose level. As specified in the protocol for cases of febrile neutropenia, the AEs resolved and the patients’ doses of MK-5108 were reduced.. Due to the observation of 2 DLTs in Panel 2 at the MK-5108 225 mg Q12hr dose level, the next lowest dose group (i.e., MK-5108 150 mg Q12hr CTl) was expanded per protocol to enroll 6 patients for a more thorough examination of the toxicity profile of this dose. None of the patients receiving MK-5108 150 mg Q12hr CT experienced any DLTs in this study. As a result, the MTD of MK-5108 CT was established at 150 mg Q12hr (300 mg/day).
Pharmacokinetic analysis
Mean plasma concentration of MK-5108 showed a biphasic decline with mean t1/2 ranging from 6.6 to 13.5 h (Fig. 1, Table 5). Median Tmax was approximately 1 to 4 h following the first dose, and 1 to 6 h following the fourth dose. The t1/2 was variable and ranged from 5 to 29 h following Dose 4. The area under the serum concentration-time curve (AUC0-12h) and Cmax appeared to increase less than dose proportionally following the first dose, but increased roughly dose proportionally following the fourth dose from 100 to 1800 mg (Q12hr).

Mean plasma concentration-time profiles for MK-5108 following BID every-12-h oral dosing in Cycle 1 (N = 3 to 6/dose level). MT monotherapy, CT combination therapy
The minimum human PK targets for efficacy of MK-5108 with Q12hr dosing over 2 days in combination with IV docetaxel, as estimated from preclinical HeLa luc xenograft experiments performed in nude rats [9], was a daily exposure of 83 μM*hour and/or a trough serum concentration of 2–3 μM. The serum MK-5108 concentration at 12 h post dose (trough) appeared to increase roughly dose proportionally. Following the fourth dose of MK-5108 in Panel 1, a mean target trough concentration of 2 μM was observed at MT doses as low as 400–800 mg Q12hr; however, the 2 μM target concentration was not reached in all 3 patients within a given dose level until the MK-5108 MT dose was equal to or greater than 1200 mg Q12hr hours. A mean target trough concentration of 2 μM was not achieved at any of the tested MK-5108 dose levels within Panel 2.
Pharmacodynamic results
The potential effects of MK-5108 on exploratory PD biomarkers were assessed using IHC and gene expression assays conducted on pre- and post-dose surrogate tissue samples (i.e., plucked scalp hair follicles and skin punch biopsies). These assays were performed on tissue samples obtained from MT patients in Panel 1, only, including a total of 8 patients with pre and post dose expression arrays passing quality control standards.
For the IHC assays, no clear dose-related increases from baseline in pHH3 and pAurB were observed following treatment with MK-5108. Overall, the changes from baseline in pHH3 and pAurB seen in this study were generally small in magnitude and scattered around zero. The effects seen in this study were well below the increases previously observed for pHH3 in preclinical rat model experiments [11, 12].
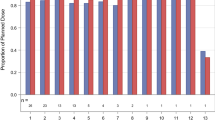
Among the seven gene expression assays tested in this study, significant dose-related changes in the expected direction were observed for 5 genes (p < 0.05 unadjusted for multiplicity; i.e., AURKA, AURKB, BIRC5, DLGAP5 [DLG7] and NDC80 [KNTC2]). Fig. 2 displays variation in the 7-gene composite score across MK-5108 dose levels. A simple linear regression test of the composite score on MK-5108 dose showed evidence of a statistically significant (p = 0.019) trend toward an increase in the composite score with increasing dose, providing evidence of target engagement in the surrogate tissue. The dose–response trend in the composite score was heavily dependent on certain individuals in the highest and lowest dose groups.

Variation in the 7-gene composite response score across MK-5108 dose levels
Tumor response
A total of 33 patients were evaluable for antitumor response in this study (Table 6). Two patients, one from each of the treatment panels, discontinued from the study due to an adverse event and hence were not included in the tumor response analysis as they had not yet completed a full treatment cycle (i.e., 1 patient receiving MK-5108 MT discontinued due to arthralgia on Day 8 and 1 patient receiving MK-5108 CT discontinued due to angioedemia on Day 9–14). There were no complete responses observed in either of the treatment panels. There was 1 partial response (5.9 %; 95 % CI: 0.1 %, 28.7 %) in a patient receiving MK-5108 225 Q12hr (450 mg/day) CT and none in the MK-5108 MT group. A total of 9 patients (50 %; 95 % CI: 26.0 %, 74.0 %) in the MK-5018 MT and 7 patients (41.2 %; 95 % CI: 18.4 %, 67.1 %) in the MK-5108 CT groups achieved stable disease. The median duration of stable disease in Panels 1 and 2 were 88 and 48 days, respectively. An additional 8 patients in each of the MK-5108 MT (44.4 %; 95 % CI: 21.5 %, 69.2 %) and MK-5018 CT (41.2 %; 95 % CI: 18.4 %, 67.1 %) groups had progressive disease.
Discussion
This study was a first-in-human, multi-center, randomized, open label, dose escalation, 2-panel phase I study investigating the safety, tolerability and spectrum of side effects associated with the AK-A inhibitor, MK-5108, administered as MT and CT with IV 60 mg/m2 docetaxel in patients with advanced and/or refractory solid tumors. In prior preclinical and clinical studies involving AKIs, single agent AKI therapy induced apoptosis in a dose- and time-dependent manner and also potentiated antitumor activity when combined with taxanes by promoting cell death by apoptosis [7, 13–15]. In preclinical studies, MK-5108 robustly potentiated apoptosis when used in combination with docetaxel in HeLaS3 cells and also enhanced the anti-tumor efficacy of docetaxel in both HeLa-luc (human cervical adenocarcinoma) and docetaxel-resistant ES-2 (human ovarian carcinoma) xenograft rat tumors without increasing docetaxel-induced hematopoietic toxicity [9]. Hence this phase I study was designed to determine the MTDs and DLTs of MK-5108 MT and CT with docetaxel in human patients with advanced and/or refractory solid tumors.
This study was terminated early by the study sponsor due to observed toxicities in Panel 2 at low doses of MK-5108 when administered in combination with docetaxel. The observed toxicities (i.e., febrile neutropenia and infection) observed at the MK-5108 225 mg Q12hr (450 mg/day) dose level + docetaxel in Panel 2 of the study exceeded the maximal allowed pre-specified threshold for DLTs. As a result, further dose escalation of MK-5108 did not continue in the CT panel which prevented achievement of the minimum projected MK-5108 PK exposure target. The minimum anticipated human PK target associated with preclinical therapeutic efficacy of MK-5108, as estimated from preclinical HeLa-luc xenograft experiments performed in nude rats was a daily exposure of 83 μM*hour and/or a trough serum concentration of 2–3 μM. This minimum PK exposure target was not attained at any of the tested MK-5108 dose levels (i.e., 100, 150, 225 mg Q12hr) in Panel 2.
The MTD of MK-5108 CT was established at 150 mg Q12hr (300 mg/day) due to the absence of DLTs in patients receiving this treatment compared with the 2 DLTs seen at the next highest MK-5108 dose level. The incidence of febrile neutropenia and Grade 3 to 4 neutropenia in patients with metastatic cancer treated with q 3 week docetaxel is high (ranging from 3 to 20 and 19 to 48 %, respectively); however, most studies have used docetaxel doses of 75–100 mg/m2 [16]. Comparable data on toxicity with doses of 60 mg/m2 q 3 week used in a patient population similar to the one studied here is sparse. A study in metastatic breast cancer [17] comparing 3 different doses of docetaxel in second line treatment reported an incidence febrile neutropenia of 5 % and Grade 3 to 4 neutropenia of 76 % at a docetaxel dose of 60 mg/m2 q 3 week. A more recent trial, conducted in chemotherapy-naïve elderly patients with advanced non-small cell lung cancer, reported febrile neutropenia occurring in 15 % and Grade 3 to 4 neutropenia in 89 % of treated patients at a docetaxel dose of 60 mg/m2 q 3 week. Therefore, in the very small cohort of patients studied here, it is impossible to conclude whether MK-5108 significantly increases the risk of hematologic toxicity at the low doses tested.
MK-5108 MT was generally well-tolerated at all doses tested with no observed DLTs. Further escalation of the MK-5108 dose beyond 1800 mg Q12hr (2600 mg/day) was prevented in Panel 1 due to early termination of the study, hence an MTD of MK-5108 MT was not established in this study. The absence of an observable MTD for MK-5108 MT at doses up to 1800 mg Q2hr (3600 mg/day) was somewhat unexpected in this study, as the toxicity profiles of other AKIs administered as single agents have been shown to be associated with neutropenia [18]. Furthermore, the established MTDs of previously studied AKIs as single agents were observed at or below the anticipated therapeutic dose for neutropenia-related DLTs [18].
With respect to the tumor response effects of MK-5108, one patient in the MK-5108 CT group achieved a PR at the 225 mg Q12hr dose level. The proportions of patients with stable disease and progressive disease were similar across the MT and CT groups. Hence, no appreciable tumor response was evident in this small study of docetaxel-refractory patients receiving MK-5108 MT and CT with relatively low doses of docetaxel (60 mg/m2). Due to the relatively small size of this study, it is difficult to definitively conclude whether MK-5108 CT may potentiate the beneficial anti-tumor effects of low-dose docetaxel. The toxicity profile observed in the MK-5108 CT group (i.e., Grade 3 and Grade 4 febrile neutropenia and Grade 3 infection) precluded escalation of MK-5108 doses to levels anticipated to provide adequate therapeutic exposure. Despite compelling preclinical findings showing the enhanced anti-tumor effects of AKIs and cytotoxic chemotherapies, it has been challenging to develop tolerable CT regimens because of the overlapping hematologic toxicities associated with both agents [18–20]. In the present study, the hematologic toxicity of neutropenia was observed only in the CT arm with docetaxel. Thus further exploration of the potential anti-tumor effects of Aurora A-specific AKIs combined with an alternative taxane dosing regimen (perhaps with granulocyte colony stimulating factor [GSCF] support or different taxane) and/or chemotherapeutic agents may be warranted.
Previous studies showed evidence of AKI-induced modulation of PD targets. Preclinical studies have suggested that, pHH3 Ser28 and gene expression markers (Aurora A, Aurora B, BIRC5, PRC1, TACC3, KNTC2, and DLG7) may be potential biomarkers of maximum PD activity of MK-5108 [21]. Among the seven genes (AURKA, AURKB, BIRC5, PRC1, TACC3, DLGAP5 [DLG7], and NDC80 [KNTC2]) tested in this study, five genes (AURKA, AURKB, BIRC5, DLGAP5 [DLG7], and NDC80 [KNTC2]) showed statistically significant, dose-related changes in gene expressions (p < 0.05) as well as evidence of target engagement in surrogate tissue (p = 0.019) following treatment with the highest and lowest MK-5108 doses. Alternatively, pAurB Thr232 is also considered as a potential PD marker of MK-5108 Aurora A selectivity, since phosphorylation of Histone H3 Ser28 appears to be dependent on Aurora B activity and autophosphorylation of Thr232 on Aurora B is critical for its activity [11, 12]. However, post treatment changes in the levels of pHH3 and pAurB with MK-5108 MT were not observed in our study despite the achievement of PK exposure levels associated with Aurora A inhibition in preclinical models. Alternative dosing strategies for MK-5108 may influence PD changes in markers of Aurora A inhibition. Other possible factors that may have hampered our ability to detect changes in PD biomarkers following MK-5108 treatment include the relatively small number of patients enrolled in this study as well as the non-homogenous nature of the patient population (e.g., different tumor types and stages, previous exposure to chemotherapy).
In conclusion, this study reached one of its co-primary objectives in defining the MTD for MK-5108 CT at 150 mg Q12hr (300 mg/day); however, an MTD for MK-5108 MT was not established even at the highest dose tested. In general, MK-5108 MT had an overall favorable toxicity profile compared with other AKIs. The lack of toxicity seen in the MK-5018 MT panel at doses that should have exceeded the therapeutic combination dose level may support further investigation of this drug in combination with taxanes (perhaps co-administered with GSCF support or employing different taxane dosing regimens) or with other chemotherapeutic agents. Nevertheless, no appreciable difference in anti-tumor activity was seen in patients receiving MK-5108 CT + low-dose docetaxel versus MK-5018 MT in this small study. While toxicity-related issues precluded the attainment of anticipated therapeutic dose levels in the CT panel, there was some evidence of target engagement in the MT panel as demonstrated by PD effects in surrogate tissues. Possible future areas of research may include evaluating the effects of MK-5108 on alternative PD biomarkers, such as the expression of proliferating cell nuclear antigen, Ki67, M30 and M65 in skin and plasma samples, as well as investigating inducible mutations in the targeted protein kinases that have been previously shown to be associated with the development of AKI resistance [4, 12, 22, 23].
References
Marumoto T, Zhang D, Saya H (2005) Aurora-A - a guardian of poles. Nat Rev Cancer 5:42–50
el Rifai W, Powell SM (2002) Molecular and biologic basis of upper gastrointestinal malignancy. Gastric carcinoma. Surg Oncol Clin N Am 11:273–291
Katayama H, Brinkley WR, Sen S (2003) The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev 22:451–464
Mountzios G, Terpos E, Dimopoulos MA (2008) Aurora kinases as targets for cancer therapy. Cancer Treat Rev 34:175–182
Lee W, Patel JH, Lockhart AC (2009) Novel targets in esophageal and gastric cancer: beyond antiangiogenesis. Expert Opin Investig Drugs 18:1351–1364
Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, Marumoto T, Saya H, Horii A (2005) RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res 65:2899–2905
Scharer CD, Laycock N, Osunkoya AO, Logani S, McDonald JF, Benigno BB, Moreno CS (2008) Aurora kinase inhibitors synergize with paclitaxel to induce apoptosis in ovarian cancer cells. J Transl Med 6:79
Tanaka E, Hashimoto Y, Ito T, Kondo K, Higashiyama M, Tsunoda S, Ortiz C, Sakai Y, Inazawa J, Shimada Y (2007) The suppression of aurora-A/STK15/BTAK expression enhances chemosensitivity to docetaxel in human esophageal squamous cell carcinoma. Clin Cancer Res 13:1331–1340
Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, Sakai T, Nambu T, Miyamoto M, Takahashi I, Miki S, Kawanishi N, Ohkubo M, Kotani H, Iwasawa Y (2010) MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther 9:157–166
Lyman GH, Kleiner JM (2011) Summary and comparison of myeloid growth factor guidelines in patients receiving cancer chemotherapy. Cancer Treat Res 157:145–165
Goto H, Yasui Y, Nigg EA, Inagaki M (2002) Aurora-B phosphorylates Histone H3 at serine28 with regard to the mitotic chromosome condensation. Genes Cells 7:11–17
Le LT, Vu HL, Nguyen CH, Molla A (2013) Basal aurora kinase B activity is sufficient for histone H3 phosphorylation in prophase. Biol Open 2:379–386
Lin Y, Richards FM, Krippendorff BF, Bramhall JL, Harrington JA, Bapiro TE, Robertson A, Zheleva D, Jodrell DI (2012) Paclitaxel and CYC3, an aurora kinase A inhibitor, synergise in pancreatic cancer cells but not bone marrow precursor cells. Br J Cancer 107:1692–1701
Qi W, Cooke LS, Liu X, Rimsza L, Roe DJ, Manziolli A, Persky DO, Miller TP, Mahadevan D (2011) Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol 81:881–890
Sehdev V, Katsha A, Ecsedy J, Zaika A, Belkhiri A, el Rifai W (2013) The combination of alisertib, an investigational Aurora kinase A inhibitor, and docetaxel promotes cell death and reduces tumor growth in preclinical cell models of upper gastrointestinal adenocarcinomas. Cancer 119:904–914
Engels FK, Verweij J (2005) Docetaxel administration schedule: from fever to tears? A review of randomised studies. Eur J Cancer 41:1117–1126
Harvey V, Mouridsen H, Semiglazov V, Jakobsen E, Voznyi E, Robinson BA, Groult V, Murawsky M, Cold S (2006) Phase III trial comparing three doses of docetaxel for second-line treatment of advanced breast cancer. J Clin Oncol 24:4963–4970
Dees EC, Cohen RB, von Mehren M, Stinchcombe TE, Liu H, Venkatakrishnan K, Manfredi M, Fingert H, Burris HA III, Infante JR (2012) Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res 18:4775–4784
Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, Rosello S, Andreu J, Jung J, Sanchis-Garcia JM, Piera A, Blasco I, Manos L, Perez-Fidalgo JA, Fingert H, Baselga J, Tabernero J (2012) Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 18:4764–4774
Diamond JR, Bastos BR, Hansen RJ, Gustafson DL, Eckhardt SG, Kwak EL, Pandya SS, Fletcher GC, Pitts TM, Kulikowski GN, Morrow M, Arnott J, Bray MR, Sidor C, Messersmith W, Shapiro GI (2011) Phase I safety, pharmacokinetic, and pharmacodynamic study of ENMD-2076, a novel angiogenic and Aurora kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 17:849–860
Carpinelli P, Moll J (2008) Aurora kinase inhibitors: identification and preclinical validation of their biomarkers. Expert Opin Ther Targets 12:69–80
Sardon T, Cottin T, Xu J, Giannis A, Vernos I (2009) Development and biological evaluation of a novel aurora A kinase inhibitor. Chembiochem 10:464–478
Pflug A, de Oliveira TM, Bossemeyer D, Engh RA (2011) Mutants of protein kinase A that mimic the ATP-binding site of Aurora kinase. Biochem J 440:85–93
Acknowledgments
This study was supported by Merck & Co., Inc., Kenilworth, NJ, USA. The authors wish to thank Dr. Amy O. Johnson-Levonas (Merck & Co., Inc., Kenilworth, NJ, USA) for her assistance with writing and editing this paper. In addition, the authors thank Kristen Lewis (Merck & Co., Inc., Kenilworth, NJ, USA) for her assistance with preparing this manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
The research complies with the current laws of the United States.
Funding
Funding for this study was provided by Merck & Co., Inc., Kenilworth, NJ, USA.
Disclosures
JL, DH, DM are current or former employees of Merck & Co., Inc., Kenilworth, NJ, USA and may hold stock/stock options in the company. CP was an employee of Merck & Co., Inc. at the time of the study and held stock in the company; she is currently an employee of Amgen and holds stock in the company. SSK has received funding for this study from Merck & Co., Inc. AW-G has received advisory board payments from Merrimack and Pfizer and study funding from Takeda. MA, SEM, PML, MNS, ACL and LT have no conflicts of interest to disclose.
Author contributions
All authors are responsible for the work described in this manuscript. All authors were involved in at least one of the following: [conception, design, acquisition, analysis, statistical analysis, interpretation of data] and [drafting the manuscript and/or revising it for important intellectual content]. All authors provided final approval of the version to be published.
Funding statement
This study was funded by Merck & Co., Inc., Kenilworth, New Jersey, USA.
Additional information
The results described in this paper were previously published in abstract form at the 2010 American Society for the College of Oncology meeting (A phase I study of MK-5108, an oral aurora A kinase inhibitor, in both monotherapy and in combination with docetaxel in patients with advanced solid tumors, Minton SE et al. J Clin Oncol. 2010;28:e13026).
clinicaltrials.gov: NCT00543387
Rights and permissions
About this article

Cite this article
Amin, M., Minton, S.E., LoRusso, P.M. et al. A phase I study of MK-5108, an oral aurora a kinase inhibitor, administered both as monotherapy and in combination with docetaxel, in patients with advanced or refractory solid tumors. Invest New Drugs 34, 84–95 (2016). https://doi.org/10.1007/s10637-015-0306-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0306-7