
Summary
Background This phase I/II study determined the maximal tolerable dose, dose limiting toxicities, antitumor activity, the pharmacokinetics and pharmacodynamics of ruthenium compound NAMI-A in combination with gemcitabine in Non-Small Cell Lung Cancer patients after first line treatment. Methods Initial dose escalation of NAMI-A was performed in a 28 day cycle: NAMI-A as a 3 h infusion through a port-a-cath at a starting dose of 300 mg/m2 at day 1, 8 and 15, in combination with gemcitabine 1,000 mg/m2 at days 2, 9 and 16. Subsequently, dose escalation of NAMI-A in a 21 day schedule was explored. At the maximal tolerable dose level of this schedule an expansion group was enrolled of which 15 patients were evaluable for response. Results Due to frequent neutropenic dose interruptions in the third week, the 28 day schedule was amended into a 21 day schedule. The maximal tolerable dose was 300 and 450 mg/m2 of NAMI-A (21 day schedule). Main adverse events consisted of neutropenia, anemia, elevated liver enzymes, transient creatinine elevation, nausea, vomiting, constipation, diarrhea, fatigue, and renal toxicity. Conclusion NAMI-A administered in combination with gemcitabine is only moderately tolerated and less active in NSCLC patients after first line treatment than gemcitabine alone.
Similar content being viewed by others


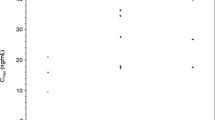
Avoid common mistakes on your manuscript.
Introduction
Ruthenium
Platinum compounds like cisplatin, carboplatin and oxaliplatin are powerful anticancer drugs, active against a variety of tumor types and widely used, but also associated with substantial side effects and primary or secondary development of resistance. [1–5] This has encouraged the search for new metal-based anti-cancer drugs with increased efficacy besides a more favorable toxicity profile and with the aim to overcome platinum resistance. [6–8] For the past decades, ruthenium (Ru), a transition metal of group 8 of the periodic table, has been considered an attractive candidate for this purpose, [9–11] because of some unique biochemical properties that theoretically might apply to ruthenium derived anti-cancer drugs when used in the human setting.
-
1)
Transferrin transport: By mimicking iron, ruthenium can bind to serum transferrin and albumin, which are proteins involved in the solubilization and transport of iron in plasma [12–15] and uptake and accumulation of ruthenium complexes is believed to be enhanced especially in tumor masses [16].
-
2)
Activation by reduction: ruthenium complexes could be considered pro-drugs: in the relative inert +3 oxidation state (Ru(III)) they are supposed to circulate almost intact in the blood, until they are reduced to the more reactive +2 oxidation state. [17] In tumor tissue, due to the more reducing environment, re-oxidation of Ru(II) to Ru(III) is less likely to occur, thus leading to an accumulation of active species. [13, 17] This would not only imply selective efficacy, but also selective toxicity [12, 13, 18].
-
3)
Slow ligand exchange kinetics: most administered metal drugs undergo spontaneous modifications prior to reaching the target (typically, some ligands are released) and therefore ligand exchange kinetics in ruthenium compounds is an important factor. Similar to platinum drugs, the ligand exchange kinetics is relatively slow (in the range of minutes to days, instead of microseconds to seconds) contributing to their general inertness and preventing rapid equilibration reactions [4, 19–21].
-
4)
DNA binding: although both platinum and ruthenium compounds bind to DNA, the binding mode differs substantially [13, 19, 22–24]. Despite intensive research, it still remains to be elucidated to what extend DNA binding is responsible for their mechanism of action [22].
NAMI-A
Imidazolium-trans-tetrachloro(dimethylsulfoxide)imidazoleruthenium(III) (C8H15Cl4N4ORu(S) or NAMI-A (acronym for Novel Anti-tumor Metastasis Inhibitor A) is the first ruthenium derived anti-cancer drug to have entered clinical evaluation [25].
NAMI-A is an imidazolium salt (replacement of Na+ by ImH+) of the earlier developed NAMI (Na[trans-RuCl4(DMSO)-(Im)]; NAMI-A is a non-hygroscopic compound with improved stability in solid state and good water solubility.
Properties, effects and proposed mechanisms of action of NAMI-A include the following:
Metastasis control: a) limiting actin dependent adhesion in vitro [26, 27]; b) limiting in vitro tumor cell motility by cytoskeleton remodeling: activation of collagen receptor integrin β1 on the cell surface results in RhoA activation and subsequently to rearrangement of the cytoskeleton in vitro.[23, 26–28]; c) anti-invasive effect in vitro and in vivo by promoting capsule formation: NAMI-A increases the extracellular matrix around tumor cells and tumor vasculature by triggering fibrotic reactions, regulates TGFβ1 expression, binds to collagen and stimulates collagen production. [29–33]; d) anti-angiogenic effect (e.g. NAMI-A inhibits in vitro the angiogenic effect induced by vascular endothelial growth factor [VEGF]) [34, 35];
It transiently blocks cell cycle progression in vitro at G2M phase. [30, 36, 37] The mechanism might be activation of Chk1, resulting in inhibition of CDC25 and subsequently in inactive phosphorylated CDC2 thereby preventing mitotic entry [23].;
In vitro it inhibits the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway and c-myc transcription [31, 38, 39];
DNA binding: although intrastrand adduct formation of NAMI-A is significantly less than of cisplatin, Ru-G and Ru-AG intrastrand adducts were observed in vitro. [40] The AG:CG adduct ratio was four times higher for NAMI-A compared to cisplatin. NAMI-A sporadically forms interstrand crosslinks, whereas the formation of DNA protein crosslinks is comparable to cisplatin [41]. Although the cytotoxic effect of NAMI-A (contrary to cisplatin) is not remarkable (on average 1,053 times less than cisplatin) [30, 36, 42], the cytotoxicity has been found to be correlated with DNA binding (which is also the case for cisplatin) [40].
Phase I study with NAMI-A in a monotherapy schedule
Prior to the study described in this article, a phase I study was performed with NAMI-A as a single agent given as an infusion over 3 h daily for 5 consecutive days every 3 weeks in patients with different types of solid tumors (including colorectal cancer, non-small cell lung cancer (NSCLC), melanoma, ovarian cancer, pancreatic cancer and mesothelioma). In total 24 patients were treated at 12 different dose levels (2.4 -500 mg/m2/day). The advised dose for further testing was 300 mg/m2/day. Adverse events included only mild hematologic toxicity, quite disabling nausea, vomiting, and diarrhea, and furthermore stomatitis, fatigue, common toxicity criteria (CTC) grade 1 and 2 creatinine increase, fever and sensitivity reactions to NAMI-A. Finally, phlebitis at the infusion site was observed when NAMI-A was administered intravenously without port-a-cath(PAC). Painful blister formation on hands, fingers and toes, although no part of the formal CTC was considered dose limiting. Twenty out of 24 patients were evaluable for response evaluation. One patient (4 %) with NSCLC experienced stable disease for 21 weeks, 19 patients (79 %) showed disease progression.
Phase I/II study with NAMI-A and gemcitabine in non-small cell lung cancer patients
Impressive pre-clinical results were observed with NAMI-A in lung tumor mouse models in which NAMI-A was especially active against tumor metastases and reduction of lung metastases was followed by increased life-expectancy. [36, 43–46] Based on the clinical results of gemcitabine in combination with platinum containing regimens in NSCLC patients, preclinical mouse studies with intravenously administered NAMI-A and gemcitabine were performed (data on file). Based on these promising preclinical results this clinical phase I study with NAMI-A and gemcitabine in NSCLC patients was initiated.
Methods
Patient selection
Patients were ≥18 years of age with confirmed histologic diagnosis of advanced NSCLC and previously treated with platinum containing therapy (i.e. cisplatin or carboplatin). All patients had an Eastern Cooperative Oncology Group performance status (ECOG-PS) of ≤2, evaluable or measurable disease according to Response Evaluation Criteria In Solid Tumors (RECIST, version 1.0) [47], a life expectancy of ≥16 weeks, adequate hepatic function defined as alanine transaminase (ALT) and aspartate aminotransferase (AST) ≤2 upper limit of normal (ULN) (≤5 times ULN in case of liver metastases), adequate renal function defined as creatinine clearance (estimated using the formula of Cockcroft and Gault) ≥50 mL/min.
Treatment plan and study design
This phase I, open label, non-randomized, dose escalation study was performed at the Netherlands Cancer Institute (NKI) in Amsterdam, the Netherlands. The study received approval of the institutional medical ethical review boards and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Written informed consent (IC) was given by all patients prior to inclusion in the study.
Initial dose escalation, in a traditional 3 + 3 design [48], was performed with NAMI-A administered as a 3 h infusion through a port-a cath at a starting dose of 300 mg/m2 at day 1, 8 and 15, in combination with gemcitabine 1,000 mg/m2 as a 30 min infusion at days 2, 9 and 16. Subsequently, dose escalation of NAMI-A in a 21 day schedule was explored: patients received NAMI-A as a 3 h port-a-cath infusion at a starting dose of 450 mg/m2 at day 1 and 8, in combination with gemcitabine 1,000 mg/m2 at days 2 and 9. The phase II part of the study consisted of expansion of the 450 mg/m2 MTD dose level of NAMI-A with gemcitabine 1,000 mg/m2 (as the 21 day schedule) by 15 evaluable patients.
Preparation of NAMI-A was consistent with the phase I study (NAMI-A monotherapy) [25].
Safety and assessments
Demographic data and medical history were collected during screening. Physical examination, vital signs and other safety assessments (ECOG-PS, registration of concomitant medication, hematology/biochemistry, urine analysis) were performed at baseline and throughout treatment: day 1, 8, 15–and 22 in the 28 day cycle–of every cycle. (During the study, based on toxicity results, the protocol was amended and the schedule changed from a 28 to 21 day cycle.)
The incidence and severity of adverse events (AEs) were evaluated and coded according to the National Cancer Institute Common Terminology Criteria (CTC) version 3.0. [49] DLT definition included: 1) any grade 3 or higher hematological or non-hematological toxicity considered to be directly related to the study drug, 2) any repeated grade 2 hematological or non-hematological toxicity considered to be directly related to the study drug and requiring dose reduction, and 3) failure to administer >75 % of the planned dosage of the study drug during cycle one as a result of treatment-related toxicity.
In case of toxicity or DLT, treatment was postponed for 1 week until recovery to CTC grade ≤1. Re-administration of study treatment occurred at a reduced dose level.
Because painful thrombophlebitis with scar formation was a known adverse event in the previously conducted phase I study with NAMI-A administered as a single agent, [25] and because the first patient in this study also developed phlebitis while NAMI-A was administered as a peripheral infusion, all additional patients in this study received NAMI-A by infusion through a port-a-cath. After a few patients were treated with antithrombotic therapy for thrombosis, it was decided (from patient 18 onwards) that all patients would receive prophylactically Fraxodi® low molecular weight heparin (LMWH) 0.3 mL daily (containing 19,000 IU anti-Xa/mL) as a subcutaneous injection.
Pharmacokinetics/pharmacodynamics
Regular blood sampling was performed to assess the pharmacokinetics of NAMI-A and gemcitabine. For NAMI-A pharmacokinetic analysis blood samples of 4 mL venous blood were collected in sodium-heparin tubes on day 1 at pre-dose, 1.5 h after start of infusion, end of infusion (EOI), EOI +15 min, EOI +30 min, 4, 6 and 8, 24 and 48 h after start of infusion. Plasma was obtained by immediate centrifugation (10 min at 4 °C, 1,500 g). Part of the plasma was transferred directly to a Centrifree® UF device with an Ultracel YM-T membrane filter with 30,000 Da molecular weight cut off (MWCO) (Millipore® Ireland Ltd, Co.Cork, Ireland) and centrifuged for 30 min at room temperature (RT) 1,500 g. The resulting plasma ultrafiltrate (pUF), representing the non-protein bound Ru fraction, and the plasma representing the total Ru concentration, were stored at −80 °C until analysis. Total and unbound Ru were determined using a validated Zeeman atomic absorption spectrometry method. Graphite furnace atomic absorption spectrometry (GF-AAS) with Zeeman correction actually measures ruthenium, but with the use of NAMI-A calibration curves the concentrations of total and unbound Ru concentrations are presented as NAMI-A levels in plasma and ultrafiltrates, respectively [50].
To analyze plasma concentrations of gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) and metabolite 2′,2′-difluorodeoxyuridine (dFdU), blood samples of 4 mL venous blood were collected in sodium-heparin tubes on day 1 at pre-dose, EOI, EOI +30 min, 2, 4, and 6 h after EOI, and 24 h after start of infusion). Immediately after sampling, blood was transferred to propylene tubes containing 0.03 mL Calbiochem® tetrahydrouridine (THU), a potent competitive inhibitor of CDA. Plasma was obtained by immediate centrifugation (5 min; 1,600x g). Analytes were quantified using validated liquid chromatography tandem mass spectrometry (LC-MS/MS) [51].
Intracellular levels of gemcitabine triphosphate (dFdC-TP), the active metabolite of gemcitabine, were determined by LCMS/MS in peripheral blood mononuclear cells (PBMCs), which were isolated from venous blood samples collected in 4 mL sodium-heparin tubes on day 1 at pre-dose, EOI, EOI +2 h according to a method previously described. [52] In short, buffy coats were collected from whole blood, and PBMC’s were separated using Ficoll-Paque™ PLUS density gradient (GE Health Care Life Sciences, UK). A volume of 10 μL of cell suspension was used for the analysis of protein concentrations. The remainder of the cell suspension was used for protein precipitation by HClO4 and extraction of the acid soluble dFdCTP. The amount of protein was used for the calculation of dFdCTP concentration in nanograms per milligram of protein.
Tumor response
Radiological disease assessments were performed by computed tomography (CT) scan or magnetic resonance imaging (MRI) at baseline and every 2 cycles, i.e. every 8 or 6 weeks. Tumor response was evaluated using RECIST 1.0. [53] Although CT-scans were performed in all patients to evaluate response to treatment, per protocol, anti-tumor activity assessment was limited to patients in the expansion cohort (in which patients were treated with the MTD of NAMI-A plus gemcitabine). In the dose expansion cohort 15 patients were required with tumors that could be evaluated by Response Evaluation Criteria In Solid Tumors (RECIST) version 1.0. Patients with tumors that could not be evaluated according to the RECIST criteria were declared not evaluable for response evaluation.
Statistical analyses
The primary endpoint of the phase I part of the study was to establish the optimal dose of the combination of NAMI-A and gemcitabine for second line treatment of NSCLC. Secondary objective was to assess the pharmacokinetic profile of the combination of NAMI-A and gemcitabine and measuring the active gemcitabine metabolite dFdC-TP concentrations in PBMCs.
The primary endpoint of the phase II part of the study was to assess the antitumor activity and secondary endpoints were the safety, DLT and MTD of NAMI-A in combination with gemcitabine in patients with NSCLC after first line treatment.
The study is a phase I dose escalation study followed by a dose expansion with a Simon two-stage design with a stopping rule [54], implicating that the first (phase I) stage of the study was designed to establish the best tolerated dose of the treatment combination in both the 21 and 28 day schedules, while in the second (phase II) stage patients were treated with the MTD defined in the first stage in order to collect activity data and additional information about the toxicity profile. The second stage, consisted of an initial 15 patients treated at the MTD. Based on response, the cohort could be expanded with 12 additional patients to a total of 27 patients. Expansion would occur in case of at least 1 response at the MTD cohort (activity of 5 % or more). Prior to the study a cutoff point of at least 15 % response rate in the second stage of the study obtained with this treatment administered at the MTD (in a group of 27 patients) was considered mandatory to consider this treatment of interest for further use: with 3 or less responses the treatment would be declared of insufficient activity, while with 4 or more responses the study would be declared of sufficient activity. This design has 80 % power to retain the treatment as active if the response rate associated with this treatment would be 20 % or more. Simon’s minimax design has been used with parameters p0 = 0.05, p1 = 0.20, alpha = 0.05 and beta = 0.20.
Results
Patients
A total of 32 patients were included in the study. One patient included in the 450 mg/m2 NAMI-A and gemcitabine 28 day dose escalation cohort never initiated study treatment, and was replaced. This patient was not included in any of the analyses. Patient characteristics are presented in Table 1. Median age of the patients was 57 years (range 40–73). The majority of patients had received 1 line of previous therapy (68 %) prior to the study and all patients had an ECOG- PS of 0 or 1. A total of 31 cycles were administered. The number of patients per dose level and the number of cycles administered are summarized in Table 2.
Safety
Thirty-one patients were evaluable for toxicity. The main treatment related adverse events per patient are presented in Table 3. In the highest dose level with 600 mg/m2 of NAMI-A, and gemcitabine administered as a 21 day cycle neutropenia grade 3 was observed in all three patients and reason for dose holds in the second week. These toxicities fulfilled the DLT criteria. In the lower 450 mg/m2 dose level of NAMI-A with gemcitabine given in a 28 day schedule, neutropenia was also frequently observed and reason for dose interruptions, especially in the third week. This observation was reason for an amended 21 day study schedule. Neutropenic dose interruptions were DLTs in the 450 mg/m2 of NAMI-A with gemcitabine 28 day schedule that declared 300 mg/m2 of NAMI-A with gemcitabine the MTD for the 28 day schedule, and in the 600 mg/m2 of NAMI-A with gemcitabine 21 day schedule that declared the 450 mg/m2 21 day schedule the MTD.
Overall, mild clinically significant hematologic toxicity occurred, mainly consisting of neutropenia (grade 2–4) and anemia (grade 2–4). Neutropenia grade 2–4 resulted in dose interruptions and dose reductions (and mostly occurred in the 600 mg/m2 of NAMI-A with gemcitabine 21 day schedule and 450 mg/m2 of NAMI-A with gemcitabine 28 day schedule).
The main non-hematological adverse events consisted of nausea or vomiting, constipation, diarrhea, transient creatinine elevation, elevated liver enzymes, and fatigue. Creatinine increase occurred in four patients (1 of 4 patients experienced a CTC grade 3 creatinine increase), all occurred at the 450 mg/m2 of NAMI-A dose levels and the increase was transient in all patients. Only in the patient with grade 3 creatinine increase short hydration was necessary. ALT and AST elevations were reason for frequent dose holds and dose reductions. Blister formation on the fingers was observed in one patient at the highest dose level consisting of 600 mg/m2 of NAMI-A and gemcitabine in a 21 day schedule.
In order to avoid phlebitis at the site of NAMI-A infusion, administration occurred by PAC in all but one patient (the first patient). Nevertheless, a significant number of patients experienced vascular/PAC related events: 2 patients with CTC grade 2 and 5 patients with CTC grade 3 (Table 3). Despite the fact that after the third patient encountering a vascular/PAC related event prophylactic use of LMWH was initiated in all patients, 4 additional patients still experienced vascular/PAC events. In all patients the vascular/PAC problems resolved completely after urokinase therapy or PAC replacement. One patient experienced a CTC grade 3 pneumonitis.
Not presented in Table 3, but observed in a significant number of patients, were transient flushing after the NAMI-A infusion, the remarkable sudden and simultaneous onset of vomiting and diarrhea, and transient change in color of urine a few hours after the NAMI-A infusion. In contrast to urine discoloration observed with KP1339 (the more soluble sodium salt of K1019), where greenish urine discoloration was observed, the color of urine turned reddish, orange and pink after NAMI-A infusion [55].
Anti-tumor activity
Anti-tumor activity was evaluated in patients in the phase II expansion cohort. In total 19 patients needed to be included to have 15 patients evaluable for response evaluation according to RECIST. [56] Four patients were not considered evaluable for response. One patient died of acute heart failure, not related to the study drugs, a week after having received one dose of both drugs. Two patients went off study prior to the first tumor evaluation due to (transients) creatinine elevation, and one patient had no tumor that was evaluable by RECIST. Of the 15 patients, 9 (60 %) experienced stable disease (SD) as best response. The other 6 (40 %) showed progressive disease (PD) after the first tumor evaluation.
In all patients that participated in the study (i.e. patients included in the phase I and II part) anti-tumor activity was observed in 15 (56 %) out of 27 patients evaluable for response, consisting of partial remission (PR) in 1 patient (4 %) and stable disease for at least 6–8 weeks in 10 patients (37 %). The patient with PR was treated at the 300 mg/m2 dose level of NAMI-A in the 21 day schedule. (See also Table 4).
Pharmacokinetics and pharmacodynamics
Blood samples for the measurement of total and unbound ultrafiltrable ruthenium, dFdC, dFdU, and dFdC-TP in PBMCs were obtained in all patients.
Figures 1a and b represent the plasma concentration time curves of total ruthenium in plasma and ultrafiltrate respectively during cycle 1 in patients receiving 300, 450 and 600 mg/m2 of NAMI-A (also for additional parameters). AUC0-48 h of bound and unbound NAMI-A was proportional to dose. Mean plasma clearance (Cl) of total and unbound NAMI-A overall dose levels was 0.31 and 64.6 L/h respectively, and the mean terminal half life over all dose levels was 61.9 and 14.1 h respectively (see Table 5 for parameters of each individual dose level). These data are all in line with the results reported in the phase I single agent study with NAMI-A [25].

a NAMI-A pharmacokinetics. Mean concentration-time curves of total NAMI-A concentration measured in plasma at different dose levels. Blue line: 300 mg/m2 NAMI-A dose level (28 day schedule), n = 3. Red line: 450 mg/m2 NAMI-A dose level (28 and 21 days schedule), n = 25. Green line: 600 mg/m2 NAMI-A dose level (21 days schedule), n = 3. In all dose levels, gemcitabine was administered at a fixed dose of 1,000 mg/m2. b NAMI-A pharmacokinetics. Mean concentration-time curves of unbound NAMI-A concentration measured in ultrafiltrates at different dose levels. Blue line: 300 mg/m2 NAMI-A dose level (28 day schedule), n = 3. Red line: 450 mg/m2 NAMI-A dose level (28 and 21 days schedule), n = 25. Green line: 600 mg/m2 NAMI-A dose level (21 days schedule), n = 3. In all dose levels, gemcitabine was administered at a fixed dose of 1,000 mg/m2
Gemcitabine (dFdC) and metabolite dFdU time-concentration curves and parameters are presented in Fig. 2 and Table 6. Gemcitabine pharmacokinetics is not altered by co-administration of NAMI-A the previous day.

Gemcitabine pharmacokinetics. Mean concentration-time curves of gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) and metabolite 2′,2′-difluorodeoxyuridine (dFdU) in plasma. In all different NAMI-A dose levels, gemcitabine was administered at a fixed dose of 1,000 mg/m2
Active gemcitabine metabolite dFdC-TP concentration-time curves of the individual patients (n = 28) measured in PBMC lysates as dFdC-TP concentrations in ng per mg of protein are presented in Fig. 3 and show a wide variability. Although sparse sampling has been performed (pre-dose, EOI +30 min, 2 h after start), mean Cmax of 2,500 ng dFdC-TP in PBMC lysate per mg of protein occurred after approximately 1 h, and lower values were found after 2 h. Overall, all values are higher than expected. Since the dFC-TP values are correlated to the protein fraction (amount of dFdC-TP is expressed per amount of protein), low protein values will directly result in high dFdCTP values. The reason for the wide variability with many low values in the protein values is unclear at present. One option that should be investigated is loss of cells due to clotting during the preparation process. Clotting might be caused by processing the samples at low temperature and/or the use of ice cold phosphate buffered saline (PBS) and cold Ficoll-Paque™ PLUS. Varying the temperature has resulted in better protein levels (data on file) but should be looked into deeper. A reason in the processing is at this stage considered most likely, especially since plasma concentrations of dFdC and dFdU seem to be in the expected range.

Gemcitabine triphosphate (dFdC-TP) concentration-time curves. Gemcitabine triphosphate (dFdC-TP) concentration-time curves of individual patients (n = 28). Measurements of dFdC-TP were performed in peripheral blood mononuclear cells (PBMCs)
Discussion
The criteria for expansion based on the obtained efficacy results were not met in the second stage of the study. Although one patient in the dose escalation part of the study experienced a PR (300 mg/m2 of NAMI-A 21 day schedule), no patients treated at the MTD in the expansion phase II part of the study experienced PR as a best response. In the extension cohort, at least one patient with a response was required to expand the cohort to 27 patients. As per protocol, the expansion with an additional 12 patients was therefore not performed and the treatment is declared to be insufficiently effective for further use. Overall, the efficacy was lower than could be expected of a treatment with gemcitabine alone. Addition of NAMI-A, based on preclinical results, was expected to be in favor of the combination NAMI-A/gemcitabine compared to gemcitabine as a single agent. However this was not observed in this study. Preclinically, NAMI-A has an optimal dose at which it gives the best antitumor response. That dose is not the maximum tolerated dose, but at which the ratio between the active compound and the inactive species into which it decomposes is in favor of the second. This fact might contribute to the lack of activity observed in this study. However, in the phase I study with NAMI-A as a single agent the efficacy was not very impressive either and combination with gemcitabine did not manage to improve this. The underlying mechanism for the reason that the encountered efficacy was less than expected with gemcitabine monotherapy remains unclear.
One patient in the dose escalation part (450 mg/m2 of NAMI-A 28 day schedule) experienced an unconfirmed PR with significant regression of lung lesions, however brain metastases increased. This is line with the assumption that NAMI-A does not cross the blood brain barrier [57].
The concept of ruthenium and other non-platinum metal anticancer drugs has intrigued scientists for over 25 years. NAMI-A is a ruthenium compound that has been extensively studied in the preclinic and showed very promising anti-metastatic results in several mouse models. [58] Activity was especially detected against metastases and more prominent than the effect on the primary tumor. [46] Connective tissue formation around tumor metastases was observed and considered to be an important explanation for the effect of NAMI-A. [33] Furthermore, NAMI-A exhibited preclinically a mild toxicity profile, superior to cisplatin, and was well tolerated by beagle dogs and mice [57, 59].
Successful pharmaceutical formulation enabled to apply NAMI-A in the clinic: although NAMI-A is stable in solid state, in solution the compound degrades rapidly upon increasing the pH (relative stability with an estimated loss of 2 % per hour is observed at pH 2–5) and hydrolysis of two chlorides from NAMI-A occurs within minutes at pH 7.4. A lyophilized formulation proved to be most suitable for parenteral use in the clinic. [60–64] Fluid prepared for infusion is stable for 3.5 h at room temperature when protected from light [25].
A phase I study with NAMI-A monotherapy given intravenously (as a 3 h peripheral infusion) for 5 consecutive days in a 21 day schedule showed blister formation on hands, fingers and toes to be DLTs. The MTD for this schedule was defined at 300 mg/m2/day. Other main AEs included peripheral phlebitis at the infusion site, sensitivity reactions to NAMI-A, significantly disabling nausea and vomiting, diarrhea, grade 1 and 2 renal dysfunction (defined as increased creatinine levels) and fever. A pre- and post-hydration schedule was used to minimize nephrotoxicity. A linear relationship between dose and AUC was observed for total and unbound ruthenium, and the t1/2 was 50 h (±19 h).
The toxicity profile of NAMI-A in combination with gemcitabine in this study is consistent with the single agent phase I study, with nausea, vomiting and diarrhea being the most prominent AEs. Transient nephrotoxicity (grade 3 creatinine elevation without ultrasound abnormalities) was observed in 4 patients and the reason to discontinue study treatment in 3 patients. Preclinically, accumulation of NAMI-A has been observed in collagen rich tissue (e.g. lungs), liver but also kidney, and nephrotoxicity was observed with increased creatinine, and histological lesions of glomeruli and tubuli of mice, which fully recovered within 15–30 days [32, 57, 65–67].
Fatigue 2-3 was also commonly observed, and in combination with the disabling nausea/vomiting and diarrhea, which often happened to occur simultaneously and with a sudden onset, this resulted in many patients experiencing the study as very exhausting and conflicting with the quality of life (QoL). AEs were scored by CTC grade and no QoL questionnaires were collected, which probably has underestimated the impact of the study on patients in the official study results, but is generally confirmed by the treating physicians and all the study personnel that had regular contact with the patients. The intense study schedule, of weekly administration of chemotherapy with the gemcitabine 1 day after the administrations of NAMI-A contributed to the subjective intensity of the study. Preclinical studies with gemcitabine and NAMI-A used this schedule, and it was considered that additional preclinical studies (demonstrating the safety of applying both agents the same day in the preclinic) were needed to administer both agents on the same day (data on file).
Elevated liver enzymes and neutropenia, which are commonly encountered adverse events with gemcitabine therapy, were also reported in this study. In the initial 28 day cycle neutropenia grade 3 often occurred in the third week leading to dose interruptions followed by the rest week. The 3 weeks on, 1 week off 28 day schedule in practice resulted in a 2 weeks on, 2 weeks off schedule. Therefore the protocol was amended into a 21 days schedule. In the 21 day 2 weeks on, 1 week off schedule the neutropenic events often coincided with the rest week and the bone marrow was generally sufficiently recovered to receive the next cycle. This schedule not only allowed intensified dosing but also allowed a higher dosing, reflected in the higher MTD (450 mg/m2 of NAMI-A compared to 300 mg/m2 both in combination with gemcitabine 1,000 mg/m2). In regard to elevated liver enzymes and neutropenia., the 21 day schedule is therefore considered the better, more practical, more patient friendly and therefore the preferred schedule. In regard to other encountered toxicities there no difference between the 21 and 28 day schedule was observed.
One patient with extensive experienced CTC grade 3 pneumonitis. This patient suffered from dyspnoea and rapid disease progression; both clinically and confirmed on CT scan evaluation, most likely all based on disease progression, but a partial contribution of NAMI-A could not be fully excluded. Therefore it has been included in the adverse event list as pneumonitis.
A significant number of patients experienced (multiple events of) upper extremity deep vein thrombosis (UEDTV) or PAC problems due to thrombosis or blood clot obstruction. It is unclear to what extend these events are related to NAMI-A, or if they can be fully attributed to common risk factors. Examples of well known risk factors include a PAC, immobility, cancer, advanced age, a recent transfusion, a history of thrombosis and co-morbidities like renal disease or infection. At the NKI PACs are commonly used in patients to administer chemotherapy and for blood sampling, especially when intravenous access in patients is difficult. Not all patients have metastasized disease like the patients in this study. Although no formal research has been performed, vascular/PAC events are however quite regularly encountered in the NKI, mostly in patient with risk factors. A partial effect of NAMI-A on the occurrence of vascular/PAC events in this particular group of patients seems to be possible, but further research is needed to establish the exact contribution of NAMI-A. The effect of prophylactic use of LMWH did not significantly lower the number of patients encountering vascular/PAC events. Whether this implicates a direct relationship with NAMI-A remains uncertain at this moment.
In conclusion, NAMI-A in combination with gemcitabine is only moderately tolerated according to the CTC criteria, and experienced by patients as a very intense treatment. Although NAMI-A should be considered a very elegant antimetastatic drug with a variety of mechanisms of action in the preclinic, a future role of NAMI-A as part of the arsenal of drugs available for physicians remains at present quite uncertain, due to the toxicity profile and the lack of convincing preliminary efficacy results. Nevertheless, additional trials in larger populations might be needed to be able to draw definitive conclusions, but only if a manner could be found that less toxicity and increased efficacy is to be expected.
References
Brabec V, Kasparkova J (2005) Modifications of DNA by platinum complexes. Relation to resistance of tumors to platinum antitumor drugs. Drug Resist Updat 8:131–146
Galanski M, Jakupec MA, Keppler BK (2005) Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr Med Chem 12:2075–2094
Galanski M (2006) Recent developments in the field of anticancer platinum complexes. Recent Pat Anticancer Drug Discov 1:285–295
Reedijk J (1999) Medicinal applications of heavy-metal compounds. Curr Opin Chem Biol 3:236–240
Weiss RB, Christian MC (1993) New cisplatin analogues in development. Rev Drugs 46:360–377
Bratsos I, Gianferrara T, Alessio E, Hartinger MA, Jakupec B, Keppler BK (2011) Ruthenium and other non-platinum anti-cancer compounds; in bioinorganic medicinal chemistry., pp 151-174
Giraldi T, Sava G (1981) Selective antimetastatic drugs (review). Anticancer Res 1:163–174
Ott I, Gust R (2007) Non platinum metal complexes as anti-cancer drugs. Arch Pharm (Weinheim) 340:117–126
Alessio E, Mestroni G, Bergamo A, Sava G (2004) Ruthenium anticancer drugs. Met Ions Biol Syst 42:323–351
Sava G, Pacor S, Mestroni G, Alessio E (1992) Na[trans-RuCl4(DMSO)Im], a metal complex of ruthenium with antimetastatic properties. Clin Exp Metastasis 10:273–280
Sava G, Bergamo A (1999) Drug control of solid tumour metastases: a critical view. Anticancer Res 19:1117–1124
Frasca D, Ciampa J, Emerson J, Umans RS, Clarke MJ (1996) Effects of hypoxia and transferrin on toxicity and DNA binding of ruthenium antitumor agents in hela cells. Metal-Based Drugs 3:197–209
Clarke MJ (2002) Ruthenium metallopharmaceuticals. Coord Chem Rev 232:69–93
Messori L, Vilchez FG, Vilaplana R, Piccioli F, Alessio E, Keppler B (2000) Binding of antitumor ruthenium(III) complexes to plasma proteins. Metal-Based Drugs 7:335–342
Messori L, Orioli P, Vullo D, Alessio E, Iengo E (2000) A spectroscopic study of the reaction of NAMI, a novel ruthenium(III)anti-neoplastic complex, with bovine serum albumin. Eur J Biochem 267:1206–1213
Allardyce CS, Dyson PJ (2001) Ruthenium in medicine: current clinical uses and future prospects. Platin Met Rev 45:62–69
Clarke MJ, Zhu F, Frasca DR (1999) Non-platinum chemotherapeutic metallopharmaceuticals. Chem Rev 99:2511–2534
Clarke MJ, Bitler S, Rennert D, Buchbinder M, Kelman AD (1980) Reduction and subsequent binding of ruthenium ions catalyzed by subcellular components. J Inorg Biochem 12:79–87
Antonarakis ES, Emadi A (2010) Ruthenium-based chemotherapeutics: are they ready for prime time? Cancer Chemother Pharmacol 66:1–9
Bloemink MJ, Reedijk J (1996) Cisplatin and derived anticancer drugs: mechanism and current status of DNA binding. Met Ions Biol Syst 32:641–685
Reedijk J (2003) New clues for platinum antitumor chemistry: kinetically controlled metal binding to DNA. Proc Natl Acad Sci U S A 100:3611–3616
Bergamo A, Sava G (2011) Ruthenium anticancer compounds: myths and realities of the emerging metal-based drugs. Dalton Trans 40:7817–7823
Bergamo A, Gaiddon C, Schellens JH, Beijnen JH, Sava G (2012) Approaching tumour therapy beyond platinum drugs: status of the art and perspectives of ruthenium drug candidates. J Inorg Biochem 106:90–99
Kostova I (2006) Ruthenium complexes as anticancer agents. Curr Med Chem 13:1085–1107
Rademaker-Lakhai JM, van den Bongard D, Pluim D, Beijnen JH, Schellens JH (2004) A Phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin Cancer Res 10:3717–3727
Frausin F, Scarcia V, Cocchietto M, Furlani A, Serli B, Alessio E, Sava G (2005) Free exchange across cells, and echistatin-sensitive membrane target for the metastasis inhibitor NAMI-A (imidazolium trans-imidazole dimethyl sulfoxide tetrachlororuthenate) on KB tumor cells. J Pharmacol Exp Ther 313:227–233
Sava G, Frausin F, Cocchietto M, Vita F, Podda E, Spessotto P, Furlani A, Scarcia V, Zabucchi G (2004) Actin-dependent tumour cell adhesion after short-term exposure to the antimetastasis ruthenium complex NAMI-A. Eur J Cancer 40:1383–1396
Bergamo A, Sava G (2007) Ruthenium complexes can target determinants of tumour malignancy. Dalton Trans; 1267-1272.
Gava B, Zorzet S, Spessotto P, Cocchietto M, Sava G (2006) Inhibition of B16 melanoma metastases with the ruthenium complex imidazolium trans-imidazoledimethylsulfoxide-tetrachlororuthenate and down-regulation of tumor cell invasion. J Pharmacol Exp Ther 317:284–291
Zorzet S, Bergamo A, Cocchietto M, Sorc A, Gava B, Alessio E, Iengo E, Sava G (2000) Lack of In vitro cytotoxicity, associated to increased G(2)-M cell fraction and inhibition of matrigel invasion, may predict In vivo-selective antimetastasis activity of ruthenium complexes. J Pharmacol Exp Ther 295:927–933
Debidda M, Sanna B, Cossu A, Posadino AM, Tadolini B, Ventura C, Pintus G (2003) NAMI-A inhibits the PMA-induced ODC gene expression in ECV304 cells: involvement of PKC/Raf/Mek/ERK signalling pathway. Int J Oncol 23:477–482
Sava G, Zorzet S, Turrin C, Vita F, Soranzo M, Zabucchi G, Cocchietto M, Bergamo A, DiGiovine S, Pezzoni G, Sartor L, Garbisa S (2003) Dual Action of NAMI-A in inhibition of solid tumor metastasis: selective targeting of metastatic cells and binding to collagen. Clin Cancer Res 9:1898–1905
Casarsa C, Mischis MT, Sava G (2004) TGFbeta1 regulation and collagen-release-independent connective tissue re-modelling by the ruthenium complex NAMI-A in solid tumours. J Inorg Biochem 98:1648–1654
Vacca A, Bruno M, Boccarelli A, Coluccia M, Ribatti D, Bergamo A, Garbisa S, Sartor L, Sava G (2002) Inhibition of endothelial cell functions and of angiogenesis by the metastasis inhibitor NAMI-A. Br J Cancer 86:993–998
Morbidelli L, Donnini S, Filippi S, Messori L, Piccioli F, Orioli P, Sava G, Ziche M (2003) Antiangiogenic properties of selected ruthenium(III) complexes that are nitric oxide scavengers. Br J Cancer 88:1484–1491
Bergamo A, Gagliardi R, Scarcia V, Furlani A, Alessio E, Mestroni G, Sava G (1999) In vitro cell cycle arrest, in vivo action on solid metastasizing tumors, and host toxicity of the antimetastatic drug NAMI-A and cisplatin. J Pharmacol Exp Ther 289:559–564
Bergamo A, Zorzet S, Gava B, Sorc A, Alessio E, Iengo E, Sava G (2000) Effects of NAMI-A and some related ruthenium complexes on cell viability after short exposure of tumor cells. Anticancer Drugs 11:665–672
Pintus G, Tadolini B, Posadino AM, Sanna B, Debidda M, Bennardini F, Sava G, Ventura C (2002) Inhibition of the MEK/ERK signaling pathway by the novel antimetastatic agent NAMI-A down regulates c-myc gene expression and endothelial cell proliferation. Eur J Biochem 269:5861–5870
Sanna B, Debidda M, Pintus G, Tadolini B, Posadino AM, Bennardini F, Sava G, Ventura C (2002) The anti-metastatic agent imidazolium trans-imidazoledimethylsulfoxide-tetrachlororuthenate induces endothelial cell apoptosis by inhibiting the mitogen-activated protein kinase/extracellular signal-regulated kinase signaling pathway. Arch Biochem Biophys 403:209–218
Pluim D, van Waardenburg RC, Beijnen JH, Schellens JH (2004) Cytotoxicity of the organic ruthenium anticancer drug Nami-A is correlated with DNA binding in four different human tumor cell lines. Cancer Chemother Pharmacol 54:71–78
Barca A, Pani B, Tamaro M, Russo E (1999) Molecular interactions of ruthenium complexes in isolated mammalian nuclei and cytotoxicity on V79 cells in culture. Mutat Res 423:171–181
Brabec V, Novakova O (2006) DNA binding mode of ruthenium complexes and relationship to tumor cell toxicity. Drug Resist Updat 9:111–122
Sava G, Pacor S, Mestroni G, Alessio E (1992) Effects of the Ru(III) complexes [mer-RuCl3(DMSO)2Im]degrees and Na[trans-RuCl4(DMSO)Im] on solid mouse tumors. Anticancer Drugs 3:25–31
Sava G, Capozzi I, Clerici K, Gagliardi G, Alessio E, Mestroni G (1998) Pharmacological control of lung metastases of solid tumours by a novel ruthenium complex. Clin Exp Metastasis 16:371–379
Sava G, Clerici K, Capozzi I, Cocchietto M, Gagliardi R, Alessio E, Mestroni G, Perbellini A (1999) Reduction of lung metastasis by ImH[trans-RuCl4(DMSO)Im]: mechanism of the selective action investigated on mouse tumors. Anticancer Drugs 10:129–138
Sava G, Gagliardi R, Bergamo A, Alessio E, Mestroni G (1999) Treatment of metastases of solid mouse tumours by NAMI-A: comparison with cisplatin, cyclophosphamide and dacarbazine. Anticancer Res 19:969–972
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Storer BE (1989) Design and analysis of phase I clinical trials. Biometrics 45:925–937
Cancer therapy evaluation program nci common terminology criteria version 3.0 (NCI-CTCv.3.0) March 31, 2003 Publish Date 09 August 2006 http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. 9-8-2006.Ref Type: internet communication
Crul M, van den Bongard HJ, Tibben MM, van Tellingen O, Sava G, Schellens JH, Beijnen JH (2001) Validated method for the determination of the novel organo-ruthenium anticancer drug NAMI-A in human biological fluids by Zeeman atomic absorption spectrometry. Fresenius J Anal Chem 369:442–445
Vainchtein LD, Rosing H, Thijssen B, Schellens JH, Beijnen JH (2007) Validated assay for the simultaneous determination of the anti-cancer agent gemcitabine and its metabolite 2′,2′-difluorodeoxyuridine in human plasma by high-performance liquid chromatography with tandem mass spectrometry. Rapid Commun Mass Spectrom 21:2312–2322
Veltkamp SA, Hillebrand MJ, Rosing H, Jansen RS, Wickremsinhe ER, Perkins EJ, Schellens JH, Beijnen JH (2006) Quantitative analysis of gemcitabine triphosphate in human peripheral blood mononuclear cells using weak anion-exchange liquid chromatography coupled with tandem mass spectrometry. J Mass Spectrom 41:1633–1642
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van GM, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Simon R (1989) Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10:1–10
Dickson, N. R., Jones, S. F., Burris, R. K., and et.al (2011) A phase I dose-escalation study of NKP-1339 in patients with advanced solid tumors refractory to treatment. J Clin Oncol (29 suppl; abstr 2607). Ref Type: Abstract
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van GM, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Cocchietto M, Sava G (2000) Blood concentration and toxicity of the antimetastasis agent NAMI-A following repeated intravenous treatment in mice. Pharmacol Toxicol 87:193–197
Sava G, Pacor S, Bergamo A, Cocchietto M, Mestroni G, Alessio E (1995) Effects of ruthenium complexes on experimental tumors: irrelevance of cytotoxicity for metastasis inhibition. Chem Biol Interact 95:109–126
Sava G, Cocchietto M (2000) Blood levels of ruthenium following repeated treatments with the antimetastatic compound NAMI-A in healthy beagle dogs. In Vivo 14:741–744
Bouma M, Nuijen B, Jansen MT, Sava G, Bult A, Beijnen JH (2002) Photostability profiles of the experimental antimetastatic ruthenium complex NAMI-A. J Pharm Biomed Anal 30:1287–1296
Bouma M, Nuijen B, Jansen MT, Sava G, Flaibani A, Bult A, Beijnen JH (2002) A kinetic study of the chemical stability of the antimetastatic ruthenium complex NAMI-A. Int J Pharm 248:239–246
Bouma M, Nuijen B, Sava G, Perbellini A, Flaibani A, van Steenbergen MJ, Talsma H, den Bosch JJ K-v, Bult A, Beijnen JH (2002) Pharmaceutical development of a parenteral lyophilized formulation of the antimetastatic ruthenium complex NAMI-A. Int J Pharm 248:247–259
Bouma M, Nuijen B, Jansen MT, Sava G, Picotti F, Flaibani A, Bult A, Beijnen JH (2003) Development of a LC method for pharmaceutical quality control of the antimetastatic ruthenium complex NAMI-A. J Pharm Biomed Anal 31:215–228
Sava G, Bergamo A, Zorzet S, Gava B, Casarsa C, Cocchietto M, Furlani A, Scarcia V, Serli B, Iengo E, Alessio E, Mestroni G (2002) Influence of chemical stability on the activity of the antimetastasis ruthenium compound NAMI-A. Eur J Cancer 38:427–435
Magnarin M, Bergamo A, Carotenuto ME, Zorzet S, Sava G (2000) Increase of tumour infiltrating lymphocytes in mice treated with antimetastatic doses of NAMI-A. Anticancer Res 20:2939–2944
Pacor S, Zorzet S, Cocchietto M, Bacac M, Vadori M, Turrin C, Gava B, Castellarin A, Sava G (2004) Intratumoral NAMI-A treatment triggers metastasis reduction, which correlates to CD44 regulation and tumor infiltrating lymphocyte recruitment. J Pharmacol Exp Ther 310:737–744
Vadori M, Pacor S, Vita F, Zorzet S, Cocchietto M, Sava G (2012) Features and full reversibility of the renal toxicity of the ruthenium-based drug NAMI-A in mice. J Inorg Biochem 118C:21–27
Acknowledgments
We would like to thank Fondazione CRTrieste and Commissariato del Governo – Fondo Trieste for providing financial support. We are greatly indebted to Ms Lidwina Wever and Dr. O. Dalesio of the trial office of The Netherlands Cancer Institute. Finally, we are thankful for the support of G. Dastoli MD, Eudax SrL, Pavia Italy and R. Bianchi, Aquisitio S.p.A., Milan Italy.
Conflict of interest
Suzanne Leijen no conflict of interest
SjaakA. Burgers, no conflict of interest
Paul Baas, no conflict of interest
Dick Pluim, no conflict of interest
Matthijs Tibben, no conflict of interest
Erik van Werkhoven no conflict of interest
Enzo Alessio, no conflict of interest
Gianni Sava, researcher involved in discovery of NAMI-A
Jos H. Beijnen, no conflict of interest
Jan H.M. Schellens no conflict of interest
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Leijen, S., Burgers, S.A., Baas, P. et al. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest New Drugs 33, 201–214 (2015). https://doi.org/10.1007/s10637-014-0179-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-014-0179-1