
Summary
Bcl-2 family proteins are the key regulators of the intrinsic apoptotic pathway, controlling the point-of no-return and setting the threshold to engage the death machinery in response to chemical damage. Bcl-2 proteins have emerged as attractive targets for anti-cancer drug development. Navitoclax is a selective, potent, orally bioavailable, small molecule Bcl-2 inhibitor. Primary endpoints assessed the safety and pharmacokinetic (PK) interactions between navitoclax in combination with carboplatin/paclitaxel or paclitaxel alone in patients with solid tumors The study comprised two arms, one a combination of navitoclax with paclitaxel and carboplatin, the second with navitoclax and paclitaxel alone. Nineteen patients were enrolled in this study. The most frequently reported treatment-emergent AEs were alopecia (57.9 %), anemia (52.6 %), nausea (52.6 %), constipation (42.1 %), diarrhea (42.1 %), fatigue (42.1 %), neutropenia (36.8 %), thrombocytopenia (36.8 %), vomiting (31.6 %), decreased appetite (31.6 %), dehydration (26.3 %), and hypomagnesaemia (26.3 %). In the light of significant hematological and non-hematological toxicity the study was ended before de-escalation of navitoclax. Only one partial response was obtained at any dose tested, thus lowering doses could not have increased efficacy. It is the combination of toxicity with modest efficacy that led to discontinuation. No apparent PK interaction was observed between navitoclax and carboplatin or paclitaxel and the combination of navitoclax and paclitaxel had modest anti-tumor activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a multifactorial disease resulting from complex genetic and epigenetic abnormalities that disrupt the normal cycle of cellular proliferation and death [1]. Escape from apoptosis is often a hallmark of cancer cells, and is associated with chemotherapy resistance or tumor relapse [2, 3]. The intrinsic apoptotic pathway is often deregulated in tumors due to a deficiency in pro-apoptotic proteins and overexpression of anti-apoptotic proteins such as Bcl-2 [4]. Up regulation of the Bcl-2 family of proteins may inhibit cells from undergoing programmed death when exposed to cytotoxic drugs, radiation, and other pro-apoptotic stimuli [4, 5]. Therefore, inhibition of these proteins represents a rational, targeted approach to developing cancer therapeutics [6, 7]. Navitoclax (ABT-263) is an orally bioavailable, potent (Ki ≤1 nM), selective, small-molecule inhibitor of Bcl-2 and related proteins, including Bcl-xL and Bcl-w [5, 6, 8]. Clinical trial experience to date suggests a limited role for navitoclax as monotherapy, at least in solid tumors [9–12].
Navitoclax enhances the activity of chemotherapeutic agents by lowering the apoptotic threshold and potentiates the effects of cytotoxic agents in solid tumor models [13–18], thus supporting the rationale for exploration of its use in combination therapy [8]. Carboplatin is a platinum-based antineoplastic agent that disrupts DNA repair and is mainly used in the treatment of ovarian, lung, and head and neck cancers [19]. Paclitaxel is one of the most effective chemotherapeutic drugs and is mainly used to treat lung, ovarian, breast cancer, and Kaposi’s sarcoma [20, 21]. The mechanism of action of paclitaxel is to promote and stabilize microtubules and inhibit late G2 or M phases of the cell cycle, thereby causing cell death. The primary objectives of the current study were to assess the safety profile of navitoclax in combination with carboplatin/paclitaxel or paclitaxel alone, study the PK interaction between the drugs, and to determine the maximum tolerated dose (MTD) of the regimen in patients with solid tumors.
Methods
Patients
Eligible patients were ≥18 years of age, with a life expectancy ≥3 months, and were required to have a cancer type for which carboplatin and/or paclitaxel was an appropriate therapy, based on histology or cytology. Patients had measurable and/or evaluable disease as defined by Response Evaluation Criteria In Solid Tumors (RECIST [22, 25]), ECOG PS [23] ≤1, and adequate function of the bone marrow (absolute neutrophil count ≥1500/μL; platelets ≥150,000/mm3; hemoglobin ≥9.0 g/dL), renal function (serum creatinine ≤1.2 mg/dL or calculated creatinine clearance ≥60 mL/min), and hepatic function and enzymes (aspartate aminotransferase [AST], alkaline phosphatase [ALP], and alanine aminotransferase [ALT] ≤3.0 × the upper limit of normal [ULN]; bilirubin ≤1.25 × ULN). Patients with bone metastasis could have ALP ≤5.0 × ULN; patients with liver metastasis could have AST, ALP, and ALT ≤5.0 × ULN. Coagulation parameters (activated partial thromboplastin time and prothrombin time) were required to be ≤1.2 × ULN. Eligibility criteria also included no current active bleeding or thrombocytopenia-associated bleeding within 1 year, concurrent therapeutic anticoagulants, and active peptic ulcer disease or other potentially hemorrhagic esophagitis or gastritis. A history of any of the following resulted in exclusion: active immune thrombocytopenic purpura, autoimmune hemolytic anemia, or being refractory to platelet transfusions (within 1 year prior to the first dose of study drug).
Study design and treatments
This Phase I, open-label study was performed at four sites in the United States. The primary objectives of this study were to assess the safety profile of navitoclax in combination with carboplatin/paclitaxel or paclitaxel alone and the pharmacokinetic (PK) interaction between the drugs in patients with solid tumors. Secondary endpoints were to evaluate preliminary data regarding objective response rate (ORR), progression free survival (PFS), time to tumor progression (TTP), overall survival (OS), duration of overall response, and Eastern Cooperative Oncology Group (ECOG) performance status. The study was approved by the institutional review board or independent ethics committee of each participating center and complied with the International Conference on Harmonization Good Clinical Practice guidelines and applicable local regulatory requirements. All patients provided written informed consent.
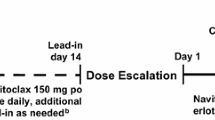
Patients were given 150 mg navitoclax orally, 30 min after the completion of breakfast, for 3 or 5 consecutive days in a 21-day cycle (Fig. 1). In the combination with carboplatin/paclitaxel, on Day 1 of each cycle, paclitaxel (175 mg/m2) was administered via IV infusion over 3 hours (immediately following navitoclax dosing), followed by IV infusion of carboplatin (AUC 4–6) over 1 hour. When navitoclax was administered in combination with paclitaxel alone, patients were treated with paclitaxel (135 mg/m2 or 175 mg/m2) via IV infusion over 3 hours on Day 1 of each cycle, immediately following navitoclax dosing. For each cycle after Cycle 1, patients had to meet the administration eligibility criteria per the paclitaxel product label (platelet count >100,000 cel/mm3 and a neutrophil count >1,500 cells/mm3) and institutional policies prior to administering chemotherapy. If the eligibility criteria were not met and chemotherapy was delayed, navitoclax administration was also delayed. The MTD was defined as the highest dose level at which fewer than 2 of 6 patients or <33 % of patients (if the cohort was expanded beyond 6 patients) experienced a DLT. Patients could continue receiving navitoclax for up to one year following the date of the last patient enrolled on study provided they completed Cycle 1 dosing of carboplatin/paclitaxel or paclitaxel alone, continued to tolerate the drug, had no evidence of disease progression, and did not meet any of the criteria for patient discontinuation.

Navitoclax (ABT-263) and carboplatin/paclitaxel dosing schematic a. navitoclax was administrated on Days 1–5 and carboplatin/paclitaxel was administrated on Day 1 in each 21-day cycle (except for Cycle 2, when navitoclax was administrated on Days 3–7 to enable PK sampling after single-agent administration) b. navitoclax was administrated on Days 1–3 and carboplatin/paclitaxel was administrated on Day 1 in each 21-day cycle (except for Cycle 2, when navitoclax was administrated on Days 3–5 to enable PK sampling after single-agent administration)
Study assessments
Safety
Investigators monitored adverse events (AEs) throughout the study. AEs were considered treatment-emergent if they occurred within 30 days of the last dose of navitoclax. Investigators rated AE severity according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 3.0 [24] or, if those criteria were not applicable, as mild, moderate, or severe. Investigators also judged the relationship of the study drugs to each AE (probably related, possibly related, probably not related, or not related). AEs were considered serious if they caused death or were life-threatening, resulted in hospitalization or prolonged a hospital stay, caused permanent disability, were congenital anomalies or abortions, or required intervention to prevent a serious outcome.
DLTs were defined as events that were attributed to navitoclax and occurred during the first cycle of concomitant dosing with navitoclax and carboplatin/paclitaxel. Any of the following events considered “possibly” related to the administration of navitoclax and met the following criteria were considered a DLT: grade 4 thrombocytopenia (platelet count <25,000/mm3); grade 2 or higher bleeding associated with thrombocytopenia; any other grade 3 AEs (except lymphopenia, leukopenia, and afebrile neutropenia; nausea, vomiting and/or diarrhea responsive to treatment within 48 hours; paclitaxel infusion-related events); all other grade 4 events (except lymphopenia, leucopenia, and afebrile neutropenia less than 7 days); and unexpected grade 2 toxicity that required dose modification or delay of >1 week. For patients with grade 2 hepatic transaminase or ALP levels at baseline as a result of liver or bone metastases, further increase to >10 × ULN was considered a DLT.
Physical examinations (including weight) were performed and vital signs measured at Screening, every week during Cycles 1 and 2 (Cycle 1 Day 1, Cycle 1 Day 8, Cycle 1 Day 15, Cycle 2 Day 1, Cycle 2 Day 8, and Cycle 2 Day 15); the first day of each subsequent Cycle (or within 72 hours prior), at the Final Visit, and at the Safety Follow-up Visit. The physical examination data collected on Cycle 1 Day 1 (predose) in the combination therapy served as the baseline physical examination for clinical assessment.
Platelet count assessments were performed and assessed by the investigator prior to study drug administration. If the platelet count on any given day was <50,000 cells/mm3, additional platelet counts were performed every day until recovery or at the discretion of the investigator. Administration of navitoclax was interrupted or discontinued for any predose platelet count <25,000 cell/mm3. The platelet count measurement obtained on Cycle 1 Day 1 (predose) in combination study served as the baseline for clinical assessment during the study.
Pharmacokinetics
Plasma samples for navitoclax assay were collected on Cycle 1 Day 1 (C1D1, navitoclax co-administrated with carboplatin/paclitaxel or paclitaxel alone) and Cycle 2 Day 3 (C2D3, navitoclax administrated alone) at pre-dose and 2, 4, 6, 8, 24 hours post dose. Plasma samples of paclitaxel were collected on C1D1 (carboplatin/paclitaxel or paclitaxel co-administrated with navitoclax) and Cycle 2 Day 1 (C2D1, carboplatin/paclitaxel or paclitaxel administrated without navitoclax) at pre-dose and 2 hours 55 min, 4, 6, 8, 24,48 hours post the start of infusion. Plasma samples of unbound platinum were collected on C1D1 (carboplatin/paclitaxel co-administrated with navitoclax) and C2D1 (carboplatin/paclitaxel administrated without navitoclax) at pre-dose and 55 min, 1.5, 2, 3, 5, 21 hours post the start of carboplatin infusion. Plasma concentrations of navitoclax and paclitaxel were determined using validated liquid chromatography methods with Tandem Mass Spectrometric detection (LC-MS/MS). Plasma concentrations of unbound platinum were determined using an inductively coupled plasma mass spectrometry. PK parameters of navitoclax, paclitaxel and unbound platinum were estimated using non-compartmental methods with Phoenix WinNonlin-Professional™, Version 6.2 (Pharsight Corporation, Mountain View, CA).
Efficacy
Tumor measurements were made according to Response Evaluation Criteria in Solid tumors (RECIST) Version 1.1 [25] at screening, the end of every second cycle, and the patient’s final visit. Disease progression was determined by the investigator based on radiologic tumor progression or clinical progression (e.g., ECOG PS ≥2, need for additional treatments, or death).
Statistical analysis
A sample size of approximately 35 patients was targeted so that the dose escalation and expanded safety portions of the study could be completed. Treatment-emergent AEs were organized by Medical Dictionary for Regulatory Activities system-organ class and preferred term, and categorized by NCI CTCAE toxicity grade and relationship to study drugs. Laboratory test results, vital signs, echocardiograms, and electrocardiogram measurements were examined for trends. Testing for PK interactions between navitoclax, carboplatin and paclitaxel were performed with repeated measures analysis. Median values and 95 % CIs for PFS, TTP, and OS were estimated with Kaplan-Meier methods. All efficacy assessments were exploratory in nature.
Results
Patients
Nineteen patients, consisting of eight males and 11 females, with a median age of 57.5 years enrolled in the study and were treated from July 7, 2009 to July 24, 2012. This resulted in an average enrollment of 0.13 patients/site/month. Eight patients received 150 mg navitoclax in combination with paclitaxel and carboplatin (2 patients followed a 5/21-day cycle and six patients followed a 3/21-day cycle), three patients received 150 mg navitoclax in combination with 175 mg/m2 paclitaxel (3/21-day cycle), and 8 patients received 150 mg navitoclax in combination with 135 mg/m2 paclitaxel (3/21-day cycle) (Fig. 1).
Data from all 19 patients were included in the demographic and safety analyses and 18 patients were included in the PK analyses. The baseline demographic characteristics of the study population are stratified by dose level in Table 1. Equal number of patients (n = 9; 47.4) had an ECOG PS of 0 or 1, 1 patient (5.3 %) had an ECOG PS of 2, at baseline. The majority of patients received ≥6 (n = 10; 52.6 %) prior therapies, including seven patients who had received carboplatin or paclitaxel (6 of the 7 received both), either as monotherapy or in combination with other agents. All patients discontinued from the study due to 1 or more causes, which included radiologically determined progressive disease (n = 13; 68.4 %), AEs (n = 3; 15.8 %), other reasons (n = 2; 10.6 %), and alternative clinical therapy (n = 1; 5.3 %).
Safety
All patients experienced at least 1 treatment-emergent AE (Table 2). The most frequently reported AEs (reported by ≥2 patients) were alopecia (57.9 %), anemia (52.6 %), nausea (52.6 %), constipation (42.1 %), diarrhea (42.1 %), fatigue (42.1 %), neutropenia (36.8 %), thrombocytopenia (36.8 %), vomiting (31.6 %), decreased appetite (31.6 %), dehydration (26.3 %), and hypomagnesemia (26.3 %). AEs patients experienced that were considered by the investigator to be possibly or probably related to navitoclax (18/19; 94.7 %) in ≥5 patients were nausea (47.4 %), fatigue (36.8 %), thrombocytopenia (36.8 %), neutropenia (31.6 %), diarrhea (31.6 %), decreased appetite (31.6 %), and alopecia (26.3 %).
A majority of patients (16/19; 84.2 %) experienced at least 1 NCI CTCAE Grade 3 or higher AE (Table 2). The most frequent reported Grade 3 or higher AEs were neutropenia (36.8 %), thrombocytopenia (36.8 %), and anemia (26.3 %). Ten (52.6 %) patients experienced at least one serious AE (SAE). The most frequently reported non-hematologic SAEs were small intestinal obstruction, dehydration, and pulmonary embolism (each reported by 2 patients, 10.5 %). Other SAEs reported included febrile neutropenia, abdominal pain, gastrointestinal ulcer, acute pancreatitis, pyrexia, hyperbilirubinemia, device-related infection, decreased appetite, hypokalemia, spinal cord compression, bronchial hemorrhage, and dyspnea. Three deaths occurred during this study, all attributed to disease progression or cancer.
Tolerability and dose-limiting toxicities
Patients were exposed to navitoclax for a median of 2.0 (range, 1–10) cycles and a median of 6.0 (range, 2–109) days. Of the 19 patients who received navitoclax, 2 (10.5 %) experienced AEs that led to discontinuation of the drug (one event of hypersensitivity in the 3/21 day schedule with carboplatin, paclitaxel and navitoclax and one of pulmonary embolism in the 5/21 day schedule with carboplatin, paclitaxel and navitoclax). Five (26.3 %) patients experienced AEs that led to a dose reduction of navitoclax. Thrombocytopenia was the most frequently reported AE that led to a dose reduction of navitoclax (n = 4; 21.1 %), interruption of navitoclax (n = 2; 10.5 %), or a delay in navitoclax dosing (n = 3; 15.8 %).
Eight patients received carboplatin (in combination with paclitaxel). Pulmonary embolism (n = 1; 12.5 %) in the 5/21 day schedule with carboplatin, paclitaxel and navitoclax, was the AE reported that led to discontinuation of the regimen. One event of neutropenia and two events of thrombocytopenia led to a dose reduction of carboplatin. Similar to navitoclax, thrombocytopenia (n = 2; 10.5 %) was the AE reported that led to a delay in carboplatin dosing.
All 19 patients received a dose of paclitaxel (eight of them in combination with carboplatin) and only one patient (treated with carboplatin/paclitaxel) experienced an AE (pulmonary embolism) in the 5/21 day schedule with carboplatin, paclitaxel and navitoclax, which led to discontinuation of the regimen. Neutropenia (n = 1; 12.5 %) and thrombocytopenia (n = 2; 10.5 %) were the AEs experienced by 2 patients that led to a dose reduction of paclitaxel. Again, thrombocytopenia was experienced by 2 (10.5 %) patients and led to a delay in paclitaxel dosing.
Five (26.3 %) patients experienced a total of 7 AEs that met the protocol-defined criteria for a DLT, including: In the 5/21-day cycle; 150 mg navitoclax + carboplatin (AUC 4–6)/paclitaxel (175 mg/m2) group, there was one event of pulmonary embolism, which lead to discontinuation of the regimen. In the 3/21-day cycle, 150 mg navitoclax + carboplatin (AUC 4–6)/paclitaxel (175 mg/m2) group, one event of pyrexia and one event of thrombocytopenia occurred. In the 3/21-day cycle; 150 mg navitoclax + paclitaxel (175 mg/m2) arm, 1 event of hyperbilirubinemia, occurred. Patients in the 3/21-day cycle; 150 mg navitoclax + paclitaxel (135 mg/m2) arm experienced hypophosphatemia, fatigue and decreased appetite as DLTs.
Pharmacokinetics
Co-administration of navitoclax with carboplatin and paclitaxel did not appear to affect the mean plasma concentration profiles of navitoclax, paclitaxel or unbound platinum (Fig. 2). The mean PK parameters of all drugs were minimally affected by the co-administration (Table 3). The data suggested that there was no significant PK interaction between navitoclax and carboplatin/paclitaxel during the co-administration.

Dose-normalized mean (+SD) plasma concentration profiles of study drugs a. navitoclax, b. paclitaxel, c. unbound platinum
Efficacy
Partial response was the best tumor response achieved by 1 (5.3 %) patient in the study who had a laryngeal invasive keratinizing squamous cell carcinoma, and was in the 3/21-day cycle; 150 mg navitoclax + paclitaxel (135 mg/m2) arm. This patient had previously achieved stable disease with carboplatin therapy. No patients achieved a complete response and 7 (36.8 %) patients had stable disease as the best response. Mean increases from baseline were observed for ECOG performance status through Cycle 6. The median PFS and TTP were 46 (n = 14; 95 % CI: 38, 134) days and the median OS was 236 (n = 3; 95 % CI could not be calculated) days. Since none of the patients achieved a CR or PR as assessed by ORR, the duration of overall response was not analyzed.
Discussion
This Phase I study evaluated safety, tolerability, PK interactions, and preliminary efficacy when navitoclax and carboplatin/paclitaxel were co-administered in adults with solid tumors. Patients began navitoclax dosing at 150 mg to mitigate navitoclax-induced thrombocytopenia since this was roughly the dose level at which the mean maximal platelet drop of approximately 60 % from baseline was observed in three different Phase I trials of navitoclax monotherapy [9, 10, 12]. In addition, administration of navitoclax for only 3 or 5 consecutive days was selected to mitigate potential overlapping toxicities between navitoclax and the cytotoxic chemotherapies, in particular the anticipated neutropenia and thrombocytopenia during the carboplatin/paclitaxel dosing regimen. However, several hematologic toxicities occurred in the 8 patients receiving navitoclax and carboplatin/paclitaxel. All 8 patients experienced at least 1 AE of Grade ≥3 with anemia (n = 4; 50.0 %), neutropenia (n = 4; 50.0 %), thrombocytopenia (n = 3; 37.5 %), and leukopenia (n = 2; 25.0 %) being the most common. This resulted in removal of carboplatin dosing from the study and it was amended to navitoclax dosing in combination with paclitaxel alone. Additional toxicities resulted in discontinuation of enrollment before the targeted 35 patients, a combination of toxicity and modest efficacy led to termination of the study.
The safety data observed in this study was consistent with the safety profile of the individual drugs (navitoclax, carboplatin, paclitaxel) administered alone. The most frequently reported AEs were alopecia, anemia, nausea, constipation, diarrhea, fatigue, neutropenia, thrombocytopenia, vomiting, decreased appetite, dehydration, and hypomagnesemia. The most frequently reported Grade ≥3 AEs were neutropenia, thrombocytopenia, and anemia. Thrombocytopenia was also the most commonly reported DLT, as well as the most commonly reported AE that led to dose reductions, dose interruptions, and dose delays of navitoclax, carboplatin, and/or paclitaxel. Small differences in the number of patients who experienced AEs were observed among the different treatment groups; however, any interpretation of these results was confounded by the small number of patients in each group.
This is the first study reporting on the combination of navitoclax with either paclitaxel or carboplatin. Navitoclax was assessed as monotherapy in several studies of patients with solid tumors, hematologic malignancies, and lymphoid cancers. Phase I and II studies in patients with small-cell lung cancer and other solid tumors found that the most frequent AEs were gastrointestinal problems, thrombocytopenia, and fatigue [9, 11]. The safety profile of navitoclax monotherapy in the previously reported studies as a group was consistent with safety findings from the present study. The MTD was not determined; the study was ended when AEs ≥ grade 3 were observed. De-escalation of navitoclax was not explored. Only one partial response was obtained at any dose tested, thus lowering doses could not have increased efficacy.
The combination of toxicity with modest efficacy led to discontinuation. Further studies might be conducted to evaluate the expression of biomarkers of the BCL2 family to as a biomarker for antitumor activities of navitoclax.
Conclusions
The MTD of navitoclax in combination with carboplatin and paclitaxel was not determined. No apparent PK interactions between navitoclax and carboplatin or paclitaxel were observed and partial response was the best objective tumor response achieved in the study.
References
Moffitt KL, Martin SL, Walker B (2010) From sentencing to execution–the processes of apoptosis. J Pharm Pharmacol 62:547–562
Fulda S (2009) Tumor resistance to apoptosis. Int J Cancer 124:511–515
Fulda S (2010) Evasion of apoptosis as a cellular stress response in cancer. Int J Cell Biol 2010:370835
Del Gaizo MV, Letai A (2008) Rational design of therapeutics targeting the BCL-2 family: are some cancer cells primed for death but waiting for a final push? Adv Exp Med Biol 615:159–175
Azmi AS, Mohammad RM (2009) Non-peptidic small molecule inhibitors against Bcl-2 for cancer therapy. J Cell Physiol 218:13–21
Kang MH, Reynolds CP (2009) Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 15:1126–1132
Leibowitz B, Yu J (2010) Mitochondrial signaling in cell death via the Bcl-2 family. Cancer Biol Ther 9:417–422
Tse C, Shoemaker AR, Adickes J et al (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68:3421–3428
Gandhi L, Camidge DR, Ribeiro de Oliveira M (2011) Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol 29:909–916
Roberts AW, Seymour JF, Brown JR et al (2012) Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 30:488–496
Rudin CM, Hann CL, Garon EB et al (2012) Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res 18:3163–3169
Wilson WH, O'Connor OA, Czuczman MS et al (2010) Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol 11:1149–1159
Ackler S, Mitten MJ, Foster K et al (2010) The Bcl-2 inhibitor ABT-263 enhances the response of multiple chemotherapeutic regimens in hematologic tumors in vivo. Cancer Chemother Pharmacol 66:869–880
Belmont L, Tan N, Wong M, et al.: Predicting synergy: Drug combination screening to identify predictive biomarkers for combination drug therapy. Mol Biol Cell 22, 2011
Chen J, Jin S, Abraham V et al (2011) The Bcl-2/Bcl-X (L)/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol Cancer Ther 10:2340–2349
Sakuma Y, Tsunezumi J, Nakamura Y et al (2011) ABT-263, a Bcl-2 inhibitor, enhances the susceptibility of lung adenocarcinoma cells treated with Src inhibitors to anoikis. Oncol Rep 25:661–667
Wong M, Tan N, Kassees R, et al.: Navitoclax enhances the activity of chemo-therapeutic and targeted agents across a large panel of epithelial cancer cell lines. Cancer Res 71, 2011
Wong M, Tan N, Zha J et al (2012) Navitoclax (ABT-263) reduces Bcl-x (L)-mediated chemo resistance in ovarian cancer models. Mol Cancer Ther 11:1026–1035
Wheate NJ, Walker S, Craig GE et al (2010) The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans 39:8113–8127
Jordan MA, Wilson L (2004) Microtubules as a target for anticancer drugs. Nat Rev Cancer 4:253–265
Voss MH, Feldman DR (2011) Paclitaxel, ifosfamide and cisplatin (TIP) beyond its original indication for salvage treatment of germ cell tumors. Onkologie 34:410–411
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, national cancer institute of the United States, national cancer institute of Canada. J Natl Cancer Inst 92:205–216
Oken MM, Creech RH, Tormey DC et al (1982) Toxicity and response criteria of the Eastern cooperative oncology group. Am J Clin Oncol 5:649–655
National Cancer Institute: Common Terminology Criteria for Adverse Events, Version 3.0, 2006
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Acknowledgments
Statistical analyses were performed by Min Tian and Joseph Beason and medical writing assistance was provided by Keith J. Gaddie, Ph.D.; all are AbbVie employees.
Disclosures
The design, study conduct, and analysis of the clinical trial were provided by AbbVie. AbbVie provided the financial support and participated in the interpretation of data, review, and approval of the manuscript.
Conflict of interest
Gordana Vlahovic is on the speaker bureau for Genentech and Pfizer. Vassiliki Karantza, Ding Wang, and David Cosgrove have no conflict of interests to declare. Nikita Rudersdorf, Jianning Yang, Hao Xiong, Todd Busman, and Mack Mabry are employees and stock owners of AbbVie.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vlahovic, G., Karantza, V., Wang, D. et al. A phase I safety and pharmacokinetic study of ABT-263 in combination with carboplatin/paclitaxel in the treatment of patients with solid tumors. Invest New Drugs 32, 976–984 (2014). https://doi.org/10.1007/s10637-014-0116-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-014-0116-3