
Abstract
Purpose
Phosphatidylinositol-3-kinase I (PI3K) inhibition sensitizes a wide range of cancer cell lines to platinum/taxane-based chemotherapy. This phase I study combines buparlisib, a pan-class 1A PI3K inhibitor, with two schedules of carboplatin and paclitaxel for patients with advanced solid tumors (ClinicalTrials.gov, NCT01297452).
Methods
There were two regimens: Group 1 received carboplatin AUC 5 and paclitaxel 175 mg/m2, on day 1 of a 21-day cycle with pegfilgrastim support; Group 2 received carboplatin AUC 5 (day 1) and paclitaxel 80 mg/m2 (days 1, 8, and 15) on a 28-day cycle without growth factor support. In both groups, three dose levels of buparlisib were explored: 50, 80, and 100 mg/day. Primary endpoint was to identify recommended phase II doses of buparlisib in both groups.
Results
Thirty subjects enrolled, 16 in Group 1 and 14 in Group 2. The DLTs were elevated alkaline phosphatase (n = 1) and uncomplicated neutropenia (n = 2). The median numbers of cycles were 5 (Group 1) and 6 (Group 2). The MTDs for buparlisib were 100 mg/day in Group 1 and 80 mg/day in Group 2. Among 25 patients with measurable disease, the confirmed objective response rate was 20 % (one complete response, four partial responses). Among three patients with known loss of PTEN expression, all derived clinical benefit from treatment.
Conclusion
The addition of buparlisib to carboplatin + paclitaxel was well tolerated, and preliminary activity was notable against tumors with loss of PTEN expression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Activating molecular alterations of the phosphatidylinositol-3-kinase (PI3K) pathway are among the most common genetic lesions identified in human cancer [1]. Well-characterized molecular lesions include mutations in the PIK3CA gene encoding the p110 catalytic subunit of PI3K, and lesions that lead to loss of function of PTEN (phosphatase and tensin homologue), a central negative regulator of the pathway. However, the efficacy of PI3K inhibitor monotherapy appears to be modest, even in individuals with tumors known to harbor genetic lesions associated with pathway activation [2]. Preclinical data indicate that a novel role for PI3K inhibitors may be their ability to augment the cytotoxic effects of conventional chemotherapeutic agents, including platinum agents and taxanes [3–5].
Buparlisib (BKM120) is an orally available 2,6-dimorpholino pyrimidine derivative that potently inhibits all class IA PI3K paralogues (p110α, β, and δ) [6]. In a first-in-human phase study, the maximum tolerated dose (MTD) of buparlisib monotherapy was determined to be 100 mg/day [7]. Among 83 patients treated in the dose escalation and expansion parts of the study, the most common adverse events were decreased appetite, diarrhea, nausea, hyperglycemia, and rash [8]. Four patients experienced radiographic responses with the following diagnoses: triple-negative breast cancer (confirmed), parotid gland carcinoma, epithelioid hemangioendothelioma, and ER + breast cancer (all unconfirmed) [7, 8].
We conducted a single-center study that was comprised of two parallel dose escalations distinguished by the carboplatin and paclitaxel schedule. The primary aim was to establish MTDs for buparlisib when given with two schedules of carboplatin and paclitaxel. The Group 1 regimen consisted of carboplatin AUC 5 and paclitaxel 175 mg/m2, both on day 1 of a 21-day cycle, with mandatory pegfilgrastim support. The Group 2 regimen was carboplatin AUC 5 (day 1) and paclitaxel 80 mg/m2 (days 1, 8, and 15) on a 28-day cycle. Prophylactic growth factor support was not used in Group 2 during the monitoring period for dose-limiting toxicity (DLT), because safety could be ensured by holding weekly chemotherapy in the event of neutropenia. Three doses of daily buparlisib were explored in each group: 50, 80, and 100 mg/day administered on a continuous basis.
Patients and methods
Eligibility criteria
The study population was derived from patients with advanced solid tumors referred for consideration of phase I trials in the Developmental Therapeutics Clinic of Memorial Sloan Kettering Cancer Center. Eligible patients had received no more than two prior cytotoxic chemotherapy regimens for recurrent and/or metastatic disease. Laboratory evidence of adequate function of bone marrow, liver, and kidneys was required. Exclusion criteria included prior treatment with a PI3K inhibitor, untreated brain metastases, history of major depressive episode or other significant psychiatric history, mood rating score of ≥10 on PHQ-9 [9] and/or ≥15 of GAD-7 [10], uncontrolled diabetes, ≥grade 2 diarrhea, prior whole pelvic radiation therapy, current use of strong inhibitors or inducers of CYP3A or QT-prolonging medications, or any uncontrolled medical conditions that could compromise participation in the study.
Primary objective
This was an open-label single-institution phase I study that was approved by the institutional review board of this hospital. The primary objectives were to determine recommended phase II doses (RP2Ds) of BKM120 given in Group 1 and in Group 2. A standard 3 + 3 dose escalation design was followed. To minimize potential confounding, Group 1 and Group 2 consents were offered to patients in an alternating fashion, without investigator or patient selection regarding group assignment.
Treatment plan and definition of dose-limiting toxicity (DLT)
Patients in both groups were evaluated by the physician in clinic and completed GAD-7 and PHQ-9 mood rating questionnaires on days 1, 8, and 15 of cycle 1, the DLT monitoring period of the study and at the start of each subsequent cycle, with additional visits as clinically indicated. Patients completed buparlisib pill diaries. Restaging imaging studies were obtained at the completion of each even-numbered cycle.
DLT was defined as any toxicity that results in treatment delay of >7 days in cycle 1, or any toxicities of grade 3 or higher (NCI Common Toxicity Criteria version 4) felt to be at least possibly related to buparlisib. Protocol-specified exceptions to this DLT definition include grade 3 hypomagnesemia, hypokalemia, or hypocalcemia if corrected within 24 h; grade 3 diarrhea lasting ≤48 h; grade 3 fatigue, nausea, vomiting, or uncomplicated hyperglycemia if resolved within 72 h; or grade 3 lymphopenia. Grade 3 hypersensitivity reaction to any of the study drugs was not deemed DLT, because such events are not strictly dose related. Uncomplicated grade 3 or 4 neutropenia lasting ≤7 days or uncomplicated grade 3 thrombocytopenia lasting ≤7 days were not considered DLTs.
Patients who remained on study after cycle 6 were offered to option to continue on protocol with buparlisib monotherapy until progression of disease or unacceptable toxicity. For patients who continued on buparlisib monotherapy after cycle 6 at a dose of <100 mg/day, it was allowable to increase to buparlisib 100 mg/day, per investigator discretion and patient preference.
Pharmacokinetic (PK) assessments
Plasma levels of buparlisib were determined from samples collected at the following time points on cycle 1/day 1: 0, 15, 30, and 60 min; 2, 3, 4.5, 6, and 8 h. On cycle 1/day 8, an additional PK blood sample was collected prior to treatment with buparlisib. Day 8, 0 h was considered as 168-h post-dose to perform the PK analysis for AUC0–168 h. Sample analysis was performed as previously described [7]. Non-compartmental analysis module in Phoenix WinNonlin® (Version 6.3) was used to assess the PK parameters of buparlisib. Peak plasma concentrations (C max) and time for the peak plasma concentrations (T max) were the observed values. The areas under the concentration time curve (AUClast) were calculated by linear trapezoidal rule. The terminal elimination rate constant was determined by regression analysis of the linear terminal portion of the log plasma concentration–time curve. Concentration–time profiles were plotted using Graphpad Prism software (Version 5).
Tissue correlative studies
Formalin-fixed paraffin-embedded (FFPE) tumor specimens, obtained as part of standard oncologic management, were subjected to mass spectrometry genotyping using the iPLEX system (Sequenom, San Diego, CA) using a multiplexed system for genotyping PIK3CA, AKT1, KRAS, NRAS, and BRAF [11–13]. Additionally, tumor PTEN expression was scored as 0, 1+, or 2+, according to previously described immunohistochemistry (IHC) methods (Dako, clone 6H2.1) [14].
Results
Baseline characteristics of the study population
Between April 5, 2011, and January 28, 2013, 30 subjects were enrolled. The data cutoff date for this analysis is May 1, 2014. Patient characteristics are summarized in Table 1. Nineteen were female and 11 were male, and median age was 53 years (range, 23–71 years). The most common tumor types were ovarian cancer (n = 5), non-small cell lung cancer (NSCLC, n = 5), nasopharyngeal cancer (n = 3), and salivary gland cancer (n = 3). Nineteen patients had received at least one prior cytotoxic chemotherapy regimen for recurrent or metastatic disease, and 16 patients had received prior radiotherapy.
Drug exposure and treatment modifications
Sixteen patients were treated in Group 1, and 14 patients were treated in Group 2. The median number of cycles administered was five in Group 1 (range <1–25 cycles) and six in Group 2 (range <1–19 cycles). Reasons for completion of ≤1 cycle of therapy were three DLTs, one withdrawal of consent, and one hypersensitivity reaction. Dose reductions and treatment delays during cycles 1 through 6 were more common in Group 2 (Supplementary Table S1). Of 24 dose reductions or treatment delays ≥7 days in Group 2, 17 were due to uncomplicated neutropenia.
Dose-limiting toxicities and recommended phase II dose in Group 1
At dose level 1 (DL1, buparlisib 50 mg/day), one patient was deemed to have experienced a DLT. She was 50 years old with NSCLC, metastatic to liver. She developed grade 3 alkaline phosphatase elevation during cycle 1, and a contributory role of study drug could not be excluded. The DL1 cohort was expanded to six patients without any other DLTs. Among three patients subsequently enrolled at DL2 (buparlisib 80 mg/day), there were no DLTs. Enrollment then began at DL3 (buparlisib 100 mg/day) in Group 1. One patient at DL3 was deemed inevaluable due to an apparent hypersensitivity reaction in cycle 1 and was replaced. There were no DLTs among the other six subjects enrolled at DL3 in Group 1. As such, buparlisib 100 mg/day was identified at the phase II recommended dose for the study drug in the Group 1 regimen.
Dose-limiting toxicities and recommended phase II dose in Group 2
There were no DLTs observed among three patients enrolled at DL1 (buparlisib 50 mg/day) or among the first three patients subsequently enrolled in DL2 (buparlisib 80 mg/day) in Group 2. Among four evaluable subjects treated at DL3 (buparlisib 100 mg/day) of Group 2, there were two DLTs (both grade 3 neutropenia lasting >7 days). A fifth subject who was enrolled at DL3 was inevaluable for the primary endpoint of the study because she erroneously self-administered BKM120 at 50 mg/day, instead of 100 mg/day as prescribed.
Because MTD was exceeded at DL3 in Group 2, three additional subjects were enrolled at DL2 (buparlisib 80 mg/day) to create a cohort of six total patients enrolled at DL2. One of these subjects withdrew consent during cycle 1 due to anhedonia that did not meet criteria for DLT, but was possibly related to buparlisib. The other two additional subjects enrolled at DL2 in Group 2 tolerated treatment well. Because five of six enrolled subjects at DL2 in Group 2 completed cycle 1 without DLT, the phase II recommended dose for buparlisib was determined to be 80 mg/day.
Table 2 summarizes the DLTs seen in both Groups 1 and 2.
Adverse event profiles of Group 1 and Group 2
Adverse events for Groups 1 and 2 are shown in Table 3. Because it is not possible to exclude fully a role for buparlisib in potentially intensifying toxicities of carboplatin and paclitaxel, adverse events are provided regardless of the investigators’ attributions of causality. Hyperglycemia, as expected, was common in both groups. Although hyperglycemia could result from both buparlisib and steroid premedications, glucose elevations were usually mild. In Group 1, ≥grade 3 adverse events were relatively uncommon, with the exception of lymphopenia. In Group 2, the incidence rates of ≥grade 3 neutropenia and leukopenia were 79 and 64 %, respectively. There were no episodes of febrile neutropenia in the study. Myalgia and arthralgia were more common in Group 1 than in Group 2, likely due to the use of pegfilgrastim in Group 1, but were usually Grade 1 or 2. There were no treatment-related deaths. Patients in Group 2, who received steroid premedication and chemotherapy more often than patients in Group 1, had a slightly lower incidence of rash (Table 3). The majority of rashes were mild and maculopapular in nature.
Pharmacokinetics
Following oral administration of buparlisib with carboplatin + paclitaxel, dose-related increases in plasma exposure to buparlisib were observed from 50 to 100 mg in both Groups 1 and 2 (Table 4). Plasma exposure (AUC0–8 h) was similar in Groups 1 and 2 at respective doses. Mean concentration–time profiles for each dose level also were similar in both Groups 1 and 2 at each dose level (Fig. 1). Individual concentration profiles of the three patients who experienced DLT were similar to those of the other patients in their respective dose levels who did not experience DLT (data not shown).

Mean concentration–time profiles of buparlisib
Efficacy
Among 30 patients, 25 had measurable disease by RECIST criteria at baseline. Best responses among patients measureable by RECIST criteria were complete response (CR) in one patient, confirmed partial response (PR) in four patients, unconfirmed partial response (uPR) in two patients, stable disease in ten patients, and progression of disease in three patients. Five of 25 patients who had measurable disease at baseline were not evaluable for response assessment due to the following events that occurred during cycle 1: DLT (n = 3), hypersensitivity reaction (n = 1), and withdrawal of consent (n = 1). Among 25 patients with measureable disease who received any treatment on study, the confirmed objective response rate (1 CR, 4 PRs) was 20 % (5/25). Among ten patients with stable disease by RECIST criteria, five patients had reductions in measurable disease of at least 20 %. Table 5A summarizes the 13 patients with measurable disease by RECIST criteria who had a best response of ≥20 % disease reduction and/or who had stable disease or better for ≥6 cycles.
Among five patients with disease that was evaluable (but not measurable) by RECIST criteria at baseline, evidence of clinical benefit with duration of at least 13 cycles was seen in each case (Table 5B). All three patients with ovarian cancer that was not measurable by RECIST criteria experienced CA-125 responses by Rustin criteria [15]. One man with metastatic parotid carcinoma to bone, with loss of PTEN expression, experienced prolonged disease control for approximately 20 months with resolution of bone pain and normalization of alkaline phosphatase (baseline 236, nadir 80; normal range 45–129 U/L).
Taken together, Table 5A and B provide clinical data for 18 patients who had reductions in measurable disease of at least 20 % and/or completed at least six cycles of treatment, comprising 60 % (18/30) of patients who received any study treatment and 72 % of patients who completed at least one cycle of study treatment (18/25).
Correlative studies of tumor tissue
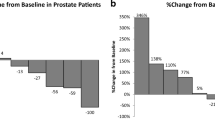
Table 5 also summarizes results of somatic mutation analysis by Sequenom and PTEN expression by IHC. Sequenom analysis was performed in 29 of 30 enrolled subjects, and PTEN IHC analysis was performed in 25 patients. There were three patients with PTEN loss (IHC score = 0; Supplementary Figure S1). Two of these patients experienced response or disease control for 14 and 19 cycles, respectively. The third patient with PTEN loss, a 60-year-old man with prostate cancer and diabetes, experienced initial reduction of measureable disease (−23 % after cycle 2). He developed disease progression at the end of cycle 4 in the context of dose reductions and delays due to worsening of diabetic control. One NSCLC patient with PIK3CA mutation (H1047R) experienced prolonged stable disease (best response, 20 % reduction) for ten cycles. Among patients who are not listed in Table 5 due to lack of clinical benefit, none were found to have PIK3CA somatic mutation or PTEN loss.
Discussion
This phase I study establishes recommended doses of oral buparlisib that can be administered with two dosing schedules of carboplatin and paclitaxel for patients with advanced solid tumors. For the Group 1 schedule (carboplatin AUC 5, paclitaxel 175 mg/m2, both on day 1 of a 21 day cycle, with pegfilgrastim support), the recommended dose of oral buparlisib is 100 mg/day, which is the recommended dose of single-agent buparlisib. For the Group 2 schedule (carboplatin AUC 5 on day 1, and paclitaxel 80 mg/m2 on days 1, 8 and 15 of a 28 day cycle, without growth factor support), the maximum tolerated dose of buparlisib was 80 mg/day. The most common DLT was uncomplicated grade 3 neutropenia in Group 2. Encouraging efficacy was seen in several tumor types, including ovarian cancer, salivary gland cancer, nasopharyngeal cancer, and non-small cell lung cancer. All three patients with tumors that harbored PTEN loss experienced objective radiographic tumor reductions or clinical benefit.
This phase I experience clearly demonstrates the safety and feasibility of adding buparlisib to carboplatin and paclitaxel. The median number of cycles administered was five in Group 1 and six in Group 2. In contrast, multiple prior studies have demonstrated marked intensification of neutropenia associated with the addition of mTOR inhibitors to cytotoxic chemotherapy [16–21].
For example, in a phase I study of daily everolimus added to bolus cisplatin and docetaxel, both given at 75 mg/m2 q21 days with mandatory pegfilgrastim support among patients with locally advanced (M0) head and neck cancer [22], it was not possible to escalate to full dose of daily everolimus due to dose-limiting neutropenia in these previously untreated patients. A definitive comparison of toxicities in the two different phase I studies cannot be done due to several variables, including the fact that myelosuppression with cisplatin plus docetaxel may differ from that of carboplatin plus paclitaxel. However, it is striking that, in the present study, it was possible to escalate to full dose daily buparlisib given with carboplatin and paclitaxel in this pretreated population, whereas it was not possible to do so with daily everolimus given with cisplatin and docetaxel in previously untreated patients who also received pegfilgrastim. As such, our impression is that the addition of buparlisib to platinum/taxane-based chemotherapy probably yields less intensification of neutropenia compared with the addition of everolimus to platinum/taxane-based chemotherapy.
In Group 2, uncomplicated grade 3 neutropenia lasting more than 7 days was the DLT that prevented escalation above buparlisib 80 mg/day. The median number of cycles for Group 2 patients indicates that neutropenia in this group generally was transient and clinically manageable.
The Group 1 regimen is attractive regimen for further clinical development in the near term, because buparlisib can be given at full dose (100 mg/daily) and dose modifications are acceptably infrequent. The Group 2 regimen also was well tolerated, but was logistically challenging to administer due to frequent dose delays (potentially, this could have been mitigated by regular use of growth factor in Group 2). In the absence of a clear signal of clinical advantage in Group 2, the Group 1 regimen appears preferable for future studies, in view of favorable tolerability and ease of administration.
Antitumor efficacy was evident in a broad range of tumor types in both groups. Among 25 patients who completed at least one cycle of therapy in Group 1 or Group 2, 18 (72 %) had reductions in measurable disease of at least 20 %, and/or completed six or more cycles. This relatively high percentage of patients experiencing clinical benefit should motivate further study of this combination in a variety of tumor types, including NSCLC, ovarian cancer, and NPC. The combination of daily buparlisib with carboplatin + paclitaxel, both given q 3 weeks, is the subject of a randomized phase II study for patients with metastatic squamous lung cancer (BASALT-2; NCT01820325).
Since the development of this study, highly potent α-selective PI3K inhibitors, such as BYL719 and GDC-0032, have become a topic of intense research interest for patients with tumors harboring hotspot PIK3CA mutations [23–25]. Pan-class I PI3K inhibitors such as buparlisib may be preferable for tumors harboring PTEN loss that are dependent on the β isoform of PI3K [26, 27]. In a xenograft model system of a PTEN-deficient tumor, synthetic lethality was achieved with the combination of buparlisib plus cisplatin [28]. The results of the current study are consistent with the notion of supra-additive efficacy when buparlisib is added to platinum-based chemotherapy, because all three patients with PTEN loss (IHC score = 0) experienced objective radiographic tumor reductions or clinical benefit. A specific question for further study is whether the Group 1 may be a highly effective option for patients with PTEN-deficient malignancies.
In summary, the current study demonstrates that the addition of buparlisib to carboplatin + paclitaxel is well tolerated when the chemotherapy is given on a q3 week cycle with pegfilgrastim support for patients with advanced solid tumors. The recommended dose of the buparlisib in the Group 1 regimen is 100 mg/day. Encouraging efficacy was seen in a broad range of solid tumor types. A topic of special interest for future study is to further describe the efficacy of buparlisib + carboplatin + paclitaxel in tumors harboring PTEN loss.
References
Courtney KD, Corcoran RB, Engelman JA (2010) The PI3K pathway as drug target in human cancer. J Clin Oncol 28:1075–1083
Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, Fu S, Piha-Paul SA, Lee JJ, Luthra R, Tsimberidou AM, Kurzrock R (2013) PIK3CA mutation H1047R is associated with response to PI3K/Akt/MTOR signaling pathway inhibitors in early phase clinical trials. Cancer Res 73:276–284
Ihle NT, Williams R, Chow S, Chew W, Beggrenm MI, Paine-Murrieta G, Minion DJ (2004) Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther 3:763–772
Brognard J, Clark AS, Ni Y, Dennis PA (2001) Akt/Protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res 61:3986–3997
Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB (2002) Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 62:1087–1092
Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann SM, Frisch C, Dorsch M, Chene P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson C, Schlegal R, Hoffman F, Garcia-Echeverria C, Sellers WR, Voliva CF (2012) Identification and characterization of NVP-BKM120, and orally available pan class I PI3-Kinase inhibitor. Mol Cancer Ther 11:317–328
Bendell JC, Rodon J, Burris HA, De Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J (2012) Phase I, dose-escalation study of BKM120, and oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 30:282–290
Rodon J, Brana I, Siu LI, de Jonge MJ, Homji N, Mills D, Di Tomaso E, Sarr C, Trandafir L, Massacesi C, Eskens F, Bendell JC (2014) Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs [Epub ahead of print]
Kroenke K, Spitzer RL, Williams JB (2001) The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 16:606–613
Spitzer RL, Kroenke K, Williams JB, Lowe B (2006) A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med 166:1092–1097
Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, Cercek A, Kemeny N, D’Angelica M, Viale A, Heguy A, Paty P, Chan TA, Saltz LB, Weiser M, Solit DB (2012) Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol 30:2956–2962
Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, Halilovic E, Wilson M, Huberman K, Ricarte-Filho JC, Persaud Y, Levine DA, Fagin JA, Jhanwar SC, Mariadason JM, Lash A, Ladanyi M, Saltz LB, Heguy A, Paty SB, Solit DB (2010) Genomic and biologic characterization of exon 4 KRAS mutations in human cancer. Cancer Res 70:5901–5911
Reidy DL, Vakiani E, Fakih MG, Saif MW, Hecht JR, Goodman-Davis N, Hollywood E, Shia J, Schwartz J, Chandrawansa K, Dontabhaktuni A, Youssoufian H, Solit DB, Saltz LB (2010) Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J Clin Oncol 28:4240–4246
Sakr RA, Barbashina V, Morrogh M, Chandarlapaty S, Andrade VP, Arroyo CD, Olvera N, King TA (2010) Protocol for PTEN expression by immunohistochemistry in formalin-fixed paraffin-embedded human breast carcinoma. Appl Immunohistochem Mol Morphol 18:371–374
Rustin G, Quinn M, Thigpen T, du Bois A, Pujade-Lauraine E, Jakobsen A, Eisenhauer E, Sagae S, Greven K, Vergote I, Cervantes A, Vermorken J (2004) Re: new guidelines to evaluate response to treatment in solid tumors (ovarian cancer). J Natl Cancer Inst 96:487–488
Andre F, Campone M, O’Regan R, Manlius C, Massacesi C, Sahmoud T, Mukhopadhyay S, Soria J-C, Naughton M, Hurvitz SA (2010) Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol 28:5110–5115
Campone M, Levy V, Bourbouloux E, Riguad DB, Bootle D, Dutreix C, Zoellner U, Shand N, Calvo F, Raymond E (2009) Safety and pharmacokinetics of paclitaxel and the oral mTOR inhibitor everolimus in advanced solid tumors. Br J Cancer 100:315–321
Kollmannsberger C, Hirte H, Siu LL, Mazurka J, Chi K, Elit L, Walsh W, Sederias J, Doyle A, Eisenhauer EA, Oza AM (2012) Temsirolimus in combination with carboplatin and paclitaxel in patients with advanced solid tumors: a NCIC-CTG, phase 1, open-label dose-escalation study (IND 179). Ann Oncol 23:238–244
Moulder S, Gladish G, Ensor J, Gonzalez-Angulo AM, Cristofanillo M, Murray JM, Booser D, Giordano SH, Brewster A, Moore J, Rivera E, Hortobagyi GN, Tran HT (2012) A phase 1 study of weekly everolimus (RAD001) in combination with docetaxel in patients with metastatic breast cancer. Cancer 118:2378–2384
Ramalingam SS, Harvey D, Saba N, Owonikoko TK, Kauh J, Shin DM, Sun S-Y, Strychor S, Tighiouart M, Egorin M, Fu H, Khuri FR (2010) Phase I and pharmacokinetic study of everolimus, a mammalian target of rapamycin inhibitor, in combination with docetaxel in recurrent/refractory nonsmall cell lung cancer. Cancer 116:3903–3909
Fury MG, Sherman E, Ho A, Katabi N, Sima C, Kelly KW, Nwankwo O, Haque S, Pfister DG (2012) A phase I study of temsirolimus + carboplatin + paclitaxel for patients with recurrent or metastatic (R/M) head and neck squamous cell cancer (HNSCC). Cancer Chemother Pharmacol 70:121–128
Fury MG, Sherman EJ, Ho AL, Xiao H, Tsai F, Nwankwo OG, Sima CS, Heguy A, Katabi N, Haque S, Pfister D (2013) A phase I study of everolimus + docetaxel + cisplatin as induction chemotherapy for patients with locally and/or regionally advanced head and neck cancer. Cancer 119:1823–1831
Rodon J, Juric D, Gonzalez-Angulo A-M, Bendell J, Berlin J, Bootle D, Gravelin K, Huang A, Derti A, Lehar J, Wuerthner J, Boehm M, van Allen E, Wagle N, Garraway LA, Yelensky R, Stephens PJ, Miller VA, Schlegel R, Quadt C, Baselga J (2013) Towards defining the genetic framework for clinical response to treatment with BYL719, a PI3K alpha specific inhibitor. Cancer Res 73 (8 Suppl. 1): Abstract LB-65
Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffman A, Guthy D, Erdmann D, De Pover A, Furet P, Gao H, Ferretti S, Wang Y, Trappe J, Brachmann SM, Maira SM, Wilson C, Boehm M, Garcia-Echeverria C, Chene P, Wiesmann M, Cozens R, Lehar J, Schlegel R, Caravatti G, Hoffman F, Sellers WR (2014) Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of patient stratification for clinical trials. Mol Cancer Ther 13:1117–1129
Ndubaku CO, Heffron TP, Staben ST, Baumgardner M, Blaquiere N, Bradley E, Bull R, Do S, Dotson J, Dudley D, Edgar KA, Friedman LS, Goldsmith R, Heald RA, Kolesnikov A, Lee L, Lewis C, Nannini M, Nonomiya J, Pang J, Price S, Prior WW, Salphati L, Sideris S, Wallin JJ, Wang L, Wei B, Sampath D, Olivero AG (2013) Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1.4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a beta-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor efficacy. J Med Chem 56:4597–4610
Wee S, Wiederschain D, Maira S-M, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao Y-M, Lengauer C (2008) PTEN deficient tumors depend of PIK3CB. Proc Natl Acad Sci USA 105:13057–13062
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ (2008) Essential roles of PI(3)K-p110beta in cell growth, metabolism, and tumorigenesis. Nature 454:776–779
Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V (2013) Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 341:395–399
Acknowledgments
They also thank Saiprasad Boddu, of Sai Life Sciences, for pharmacokinetic analyses. ClinicalTrials.gov identifier: NCT01297452. The study site received funding from Novartis Pharmaceuticals.
Conflict of interest
M.F., A.H., R.C., M.L, and M.V. have served on advisory boards and/or consulted for Novartis. No potential conflict of interest was disclosed by the other authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Clinicaltrials.gov ID NCT01297452.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hyman, D.M., Snyder, A.E., Carvajal, R.D. et al. Parallel phase Ib studies of two schedules of buparlisib (BKM120) plus carboplatin and paclitaxel (q21 days or q28 days) for patients with advanced solid tumors. Cancer Chemother Pharmacol 75, 747–755 (2015). https://doi.org/10.1007/s00280-015-2693-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2693-z