
Summary
The novel compound N-benzoxazol-2-yl-N′-1-(isoquinolin-3-yl-ethylidene)-hydrazine (EPH136) has been shown to exhibit antitumor activity in vitro and in vivo. A COMPARE analysis showed that the patterns of cellular effects of EPH136 are not related to any of 175 standard antitumor agents with a known mechanism of action. In order to help identify the mechanism of action we employed a bioinformatics approach called partial least squares modelling in latent variables in which the expression levels of ~8,000 genes in each of 56 untreated NCI panel cell lines were correlated with the respective IC50 values of each cell line following treatment with EPH136. The 60 genes found to be most important for the antiproliferative effect of EPH136 are involved in nucleoside, nucleotide, nucleic acid binding and metabolism, developmental processes, protein modification and metabolism. In addition, using a DNA microarray we measured the expression of ~5,000 known genes following treatment of HT-29 colon carcinoma cells with a two-fold IC50 concentration of EPH136. The genes that were up-regulated more than two-fold compared to untreated controls belong to the same classes as found by the bioinformatic approach. Many of these proteins are regulated by oxidation/reduction and so we concluded that formation of radicals may be involved in the mechanism of action. We show here that EPH136 leads to generation of oxygen radicals, swelling of mitochondria and dissipation of the mitochondrial membrane potential. The antiproliferative activity of EPH136 was prevented by the radical scavenger N-acetylcysteine. Cells with elevated glutathione exhibited resistance to EPH136. In summary, the mechanism of the novel experimental anticancer drug EPH136 is generation of radicals and dissipation of the mitochondrial membrane potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Novel approaches for obtaining antitumor agents comprise targeting of signal transduction molecules such as bcr/abl by Gleevec [1], development of antiangiogenic compounds [2], valrubicin, a cytoplasmic targeted anthracycline interacting with protein kinase C [3] and application of antisense oligonucleotides directed against bcl2 [4] or PKCα [5]. Other perspectives include gene therapy or targeted antibodies. We synthesized novel bicyclic hydrazones which show antitumor activity against solid tumors in vitro and in vivo. The mechanism of action of these compounds is not known [6–9]. Several of these novel compounds, among them N-benzoxazol-2-yl-N′-1-(isoquinolin-3-yl-ethylidene)-hydrazine (EPH136; Fig. 1), were also screened against the panel of 60 cell lines at the National Cancer Institute (NCI). Many thousands of new compounds have been screened in this panel of cell lines, among them the so-called standard antitumor agents. The GI50 (IC50) pattern of each of these standard agents in all cell lines is well established. These activity patterns, also called “fingerprints” can be expressed as “mean graphs”. Similarity in pattern often indicates similarity in mechanism of action. The COMPARE algorithm can be used to find the mechanism of action of a “seed” compound with an unknown mechanism of action by comparison with the pattern of known compounds [10–12]. A COMPARE analysis showed that the mechanism of action of EPH136 (NSC 693638) does not match that of the standard antitumor agents and therefore exhibits a new and unknown mechanism of action [9].

Structure of N-benzoxazol-2-yl-N′-1-(isoquinolin-3-yl-ethylidene)-hydrazine (EPH136)
The NCI database also contains data on the expression levels of 9,799 cDNAs representing ~8,000 genes in the 60 cell lines (without any treatment). The GI50 patterns in connection with the gene expression data can be used for bioinformatic approaches to investigate the complex relationships between gene expression and antiproliferative activity. The data have been used to determine the mechanism of action of antitumor drugs [13] and also to find genes involved in the action of antitumor drugs such as RNA/DNA metabolites [14]. More recently, the multivariate statistical procedure “partial least squares projections to latent structures” PLS [15] revealed [16] that the translocating chain-associating membrane protein (TRAM) is among those important in determining the sensitivity to the antitumor drug 17-allylamino,17-demethoxygeldanamycin (17AAG). Indeed, silencing experiments by RNA interference confirmed that low levels of TRAM expression increased sensitivity of H226 cells to 17AAG, thus providing experimental support to bioinformatics “clues” [16]. In the present work we selected the PLS approach to find out which genes are important for the antiproliferative activity of EPH136 in order to help identify the target of this novel compound.
Sensitivity to different antitumor drugs is associated with distinct gene expression profiles [17]. Treatment of cells with an antitumor drug might lead to the expression of certain genes, the expression profile of which may be helpful in elucidating the mechanism of action of the compound. In this study, we used the microarray technology to monitor the mRNA expression profile of ~5,000 known genes in HT-29 colon cancer cells after treatment with EPH136. The bioinformatics approach to genes linked to the antiproliferative activity of EPH136, and the gene expression profile following treatment with EPH136 was employed to orient the search for the mechanism of action of the compound.
Materials and methods
Cell culture
Burkitt’s lymphoma CA46, HT-29 colon carcinoma, A2780 and A2780CP (elevated glutathione) ovarian carcinoma cells were grown in RPMI 1640 medium containing 10% (HT-29, A2780, A2780CP) or 15% (CA46) fetal calf serum, 2 mM glutamine and 50 μg/ml gentamycin. Inhibition of cell proliferation was determined by incubation of Burkitt’s lymphoma CA46 cells (105 cells/ml) in presence or in absence of N-acetylcysteine (NAC). The cells were incubated with NAC for 45 min. Subsequently, various concentrations of EPH136 were added. NAC was dissolved in culture medium (0.5 M), the pH neutralized with NaOH and added to the cells to obtain a final concentration of 10 mM. After 72 h the cell number was determined with an electronic cell counter (Casy1, Schaerfe System, Reutlingen, Germany). In order to count only viable cells, a size-gate was set to exclude dead cells. Determination of GI50 values (Fig. 2) was performed at the National Cancer Institute, Bethesda, MD.

Mean graph (log GI50 = IC50) of the NCI panel of cell lines following treatment with EPH136. Columns to the right show cell lines that are more, columns to the left cell lines that are less affected than the mean (38.9 nM, indicated as 0). Each log10 IC50 value is subtracted from the average. Thus, a bar projecting one unit to the right denotes that the IC50 for that cell line occurs at a concentration ten times less than the average concentration required for all the cell lines employed in the experiment. The following cell lines were used (order from top to bottom). Leukemia: CCRF-CEM, HL-60(TB), K-562, MOLT-4, RPMI-8226, SR. Non-small cell lung cancer: A549/ATCC, EKVX, HOP-62, HOP-92, NCI-H226, NCI-H23, NCI-H322M, NCI-H460, NCI-H522. Colon: COLO 205, HCC-2998, HCT-116, HCT-15, HT29, KM12, SW-620. CNS: SF-268 SF-295 SF-539 SNB-19 SNB-75 U251. Melanoma: MALME-3M, M14, SK-MEL-2, SK-MEL-28, SK-MEL-5, UACC-257, UACC-62. Ovarian cancer: IGROV1, OVCAR-3, OVCAR-4, OVCAR-5, OVCAR-8, SK-OV-3. Renal cancer: 786-0, A498, ACHN, CAKI-1, RXF 393, SN12C, UO-31. Prostate cancer: PC-3, DU-145. Breast cancer: MCF7, NCI/ADR-RES, MDA-MB-231/ATCC, HS 578T, MDA-MB-435, MDA-N, BT-549, T-47D
Cell cycle and apoptosis
Logarithmically growing Burkitt’s lymphoma cells were treated with concentrations of EPH136 corresponding to the four-fold IC50 for 24 or 48 h in phosphate buffered saline. To 2 ml of cells (~106 cells), 100 μl of 250 μg/ml propidium iodide containing 1% Triton X-100, and 25 μl of RNase A (10 mg/ml, Sigma Chemicals, Vienna, Austria) were added and incubated at room temperature for 1 h. Cell cycle analysis and quantification of apoptotic cells was performed by a FACscan (Becton Dickinson, Mountain View, CA) as described [18]. For detection of apoptotic cells by annexin V the Annexin-V-FLUO Staining Kit (Roche, Vienna, Austria) was used. Following treatment with EPH136 for the times indicated, 106 Burkitt’s lymphoma CA46 cells were washed in phosphate buffered saline, resuspended in 100 μl staining solution and incubated at room temperature for 15 min. Subsequently 0.4 ml of binding buffer were added and samples were analyzed by a FACscan (Becton Dickinson).
Incorporation of thymidine and cytidine into DNA, and uridine into RNA
Incorporation of [2-14C]cytidine into DNA was taken as an indicator for ribonucleotide reductase, incorporation of [5-3H]thymidine for DNA synthesis and of [2-14C]uridine for RNA synthesis. For cytidine incorporation, 3 ml (1–2.106 cells/ml) of exponentially growing Burkitt’s lymphoma cells were incubated (37°C) in medium with various concentrations of EPH136. After 90 min, 0.7 μCi [2-14C]cytidine (Moravek Biochemicals, Brea, CA) were added to each sample and incubated for another 60 min. Subsequently, the cells were washed twice with ice-cold PBS, resuspended in 1 ml ice-cold trichloroacetic acid (TCA) and transferred to Eppendorf tubes. Following an incubation period on ice for 20 min, the disrupted cells were centrifuged at 3,000 g for 5 min (4°C). The pellet was resuspended in 800 μl of 80 mM Tris–HCl (pH 8) to which 20 μl DNase-free RNase (10 mg/ml) was added. Following a 2 h incubation at 37°C, the solution was cooled on ice, and 200 μl 50% TCA was added. The suspension was incubated at 4°C overnight. TCA-precipitable material was obtained by centrifuging at 4,000 g (4°C) for 5 min. The pellet was washed four times with 1 ml of ice-cold 5% TCA and dissolved in 0.4 ml of 1.25 M sodium hydroxide overnight and counted in 2 ml scintillation fluid (Ultima Gold, Packard, Meriden, CT, USA) to evaluate the [2-14C]cytidine incorporated into DNA. The IC50 was determined by taking the untreated controls as 100%. For determining inhibition of DNA or RNA synthesis, 2 ml of 5.105/ml Burkitt’s lymphoma cells were incubated for 90 min with the indicated concentrations of EPH136. Subsequently, 1 μCi [3H]thymidine/ml or 0.05 μCi [14C]uridine/ml (Moravek Biochemicals, Brea, CA, USA), respectively, were added for 60 min. The cells were washed twice with ice-cold PBS after which 1 ml of 5% TCA was added. The TCA-precipitable material was transferred onto prewashed Whatman GF/C filters and washed four times with 5% TCA/1% tetrasodium diphosphate. The filters were counted in 4 ml Ultima Gold (Packard) in a β-counter (Packard).
Bioinformatics
The multivariate statistical procedure “partial least squares projections to latent structures” (PLS [15]) was carried out by means of SIMCA software package (SIMCA-P 8.0, Umetri AB, Umea, Sweden) using an X data matrix (descriptors) containing 548,744 (9,799 × 56) elements x ik , where index k is used for the x variables, i.e. the gene expression profiles (GC) or molecular targets (MT), index i for the objects, i.e. the cell lines, and the antitumor activities of EPH136 in each cell line, expressed as log GI50, as the dependent variable (y). The PLS model describes the X matrix by a principal component-like model (Eq. 1) and the y values as a predictive relationship with the X matrix principal components, under the constraint of maximizing the correlation between y and t (Eq. 2, where b a is a proportionality coefficient for each dimension a. The number of significant dimension, A, was determined by the SIMCA program by using the cross validation technique [15].
The statistical results obtained by the PLS method are able to detect what variables in the X block are relevant to determine the dependent variable (y) by means of the variable importance for the projection (VIP) values. The VIP values reflect, in fact, the importance of terms in the model both with respect to y, i.e. its correlation to the biological response, and with respect to X. SIMCA computes VIP values (SIMCA-P 8.0 User Guide and Tutorial, 1999, Umetri AB, Umea, Sweden) [15] by summing over all model dimensions the contributions VIN (variable influence).
For a given PLS dimension, a,\(\left( {{\text{VIN}}} \right)_{ak}^2 \) is equal to the squared PLS weight (w ak )2 of that term, multiplied by the percent explained of residual sum of squares by that PLS dimension. The accumulated (over all PLS dimensions) value is then divided by the total percent explained of residual sum of squares by the PLS model and multiplied by the number of terms in the model.
Analysis of gene expression by gene array
Total RNA of HT-29 colon carcinoma cells treated with two-fold IC50 (=80 nM) of EPH136 for 24 h and of untreated controls was isolated by RNAzol™ B (Tel-Test Inc., Friendswood, TX, USA) as described by the manufacturer. DNA-microarrays (membrane GeneFilters™, GF211, Research Genetics, Huntsville, AL, USA) containing ~5,000 known genes were employed to determine mRNA levels. cDNA synthesis with oligo-dT, [33P]-dCTP (10 mCi/ml, ICN, Costa Mesa, CA, USA), prehybridization, hybridization and washing was performed according to the manufacturer’s instructions. The radioactivity in the membrane array was detected by a STORM 2000 (Molecular Dynamics) phosphoimager and analyzed by Pathways™ software (Research Genetics).
Reactive oxygen species (ROS)
Luminol-dependent chemiluminescence indicating ROS was determined as described by Weiss et al. [19]. Various concentrations of EPH136 were added to 750 μl phosphate buffered saline and incubated at room temperature for 2 h. From a 10 mM stock solution of luminol (5-amino-2,3-dihydro-1,4-phtalazinedione, Sigma, Munich, Germany) in dimethylsulfoxide, a 40 μM working solution in phosphate buffered saline was prepared. 150 μl of the luminol working solution was added to the 750 μl. Subsequently, 200 μl 1% hydrogen peroxide in phosphate buffered saline was injected and the hydrogen-peroxide-induced luminescence was measured for 30 s (Luminat LB9501, Berthold, Bad Wildbad, Germany).
Mitochondrial membrane potential
JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′ tetraethylbenzimidazolylcarbocyanine iodide/chloride) is a cationic dye that exhibits potential-dependent accumulation in mitochondria, indicated by fluorescence emission shift from green (~525 nm) to red (~590 nm). 2.105 Burkitt’s lymphoma CA46 cells were seeded in 1 ml (in 24-well plates) and treated for 2, 24 or 48 h with 250 nM EPH136, or 10 min with 100 nM of the ionophore valinomycin (Sigma) as positive control. A stock solution of 5 mg/ml JC-1 (Molecular Probes) was dissolved in DMSO. From this stock solution a concentration of 10 μg/ml in RPMI 1640 medium containing 15% fetal calf serum at 37°C was prepared. At the end of the incubation with EPH136 or valinomycin, 1 ml medium containing 10 μg/ml JC-1 was added to 1 ml Burkitt’s lymphoma cells (final concentration of 5 μg/ml JC-1) and incubated at 37°C for 10 min. Subsequently the cells were washed three times with ice-cold phosphate buffered saline containing 0.1% bovine serum albumin, resuspended in 500 μl phosphate buffered saline (without bovine serum albumin) and analyzed by a fluorescence-activated cell sorter (FACscan, Becton Dickinson) as described by the manufacturer.
Real time live confocal imaging of mitochondria
Two fluorescent dyes, fluorescent wheat germ agglutinin (WGA, Molecular Probes, Eugene, OR, USA) and tetramethylrhodamine methyl ester perchlorate (TMRM, Sigma-Aldrich) were employed to visualize cell morphology (WGA) and mitochondrial activity (TMRM). This lipophilic cation accumulates in mitochondria by a potential driven uptake. To 400 μl CA46 Burkitt’s lymphoma cells in a 8-well chambered coverglass (Nunc) WGA to a final concentration of 10 μg/ml and TMRM to a final concentration of 100 nM were added. Following an incubation at 37°C in the dark for 15 min, confocal microscopy was performed with a microlens-enhanced Nipkow disk-based confocal system UltraVIEW RS (Perkin Elmer, Wellesey MA, USA) mounted on an Olympus IX-70 inverse microscope (Olympus, Nagano, Japan). Images were acquired using the UltraVIEW RS software (Perkin Elmer).
Results
Effects of EPH136 on proliferation, apoptosis and cell cycle
EPH136 was tested on the NCI panel of cell lines (Fig. 2). The compound inhibited 5 out of 6 leukemic, seven out of nine non-small cell lung cancer, six out of seven colon cancer, and five out of seven renal cancer cell lines at lower doses than the average. CNS, melanoma, ovarian, prostate and breast cancer cell lines showed mixed results (Fig. 2). The average IC50 over all cell lines was found to be 38.9 nM. The compound achieved a score of 24 and a net cell kill in the hollow fiber assay performed at the NCI [9], in that publication, according to the rules of the journal, EPH136 is denominated E-13k. A score above 20 or a net cell kill is considered a positive result and such compounds are referred for further testing.
Since the mechanism of action of bicyclic hydrazones might be related to that of semicarbazones which inhibit ribonucleotide reductase, we compared the induction of apoptosis and the effect on the cell cycle by EPH136 with that of the ribonucleotide reductase inhibitor hydroxyurea. Because it is known that the topoisomerase I inhibitor camptothecin induces apoptosis we employed this compound as another control. As shown in Fig. 3A, a four-fold IC50 concentration of EPH136 (144 nM) induced less apoptosis than camptothecin or hydroxyurea. Concentrations up to 1 μM of EPH136 induced apoptosis in approximately 20% of the cells (data not shown), indicating that apoptosis is not the leading mechanism of cell death. The compound also did not induce senescence as determined by β-galactosidase. Therefore, necrosis is causing cell death. This could be observed when Burkitt’s lymphoma cells were counted with an electronic cell counter with a size-gate showing dead and disintegrated cells and with propidium iodide without Triton-X100 (data not shown). We also compared the effects of a four-fold IC50 concentration of EPH136 (144 nM) on the cell cycle with those of a four-fold concentration of hydroxyurea (560 μM) and camptothecin (52 nM). Figure 3B shows that the ribonucleotide reductase inhibitor hydroxyurea led to an accumulation of cells in the S-phase, whereas a EPH136 did not induce significant alterations in the cell cycle, as was the case also with camptothecin.

A Induction of apoptosis. Burkitt’s lymphoma cells were treated with four-fold IC50 concentrations of the indicated drugs for 24 or 48 h. The percentage of apoptotic cells was determined by propidium iodide as described in “Materials and Methods” The mean (±SD) of three independent experiments, in which two samples were taken within each experiments, is shown. (Campto Camptothecin; HU hydroxyurea). B Cell cycle analysis. Burkitt’s lymphoma cells were treated with four-fold IC50 concentrations of the indicated drugs for 48 h. Cell cycle analysis was performed with propidium iodide as described in “Materials and Methods”. The percentage of cells in the different phases of the cell cycle (mean ± SD) of three independent experiments, in which two samples were taken within each experiment, is shown
Inhibition of DNA synthesis, RNA synthesis and ribonucleotide reductase
Many antitumor agents interact with DNA or nucleotide synthesis. Therefore, we investigated the effects of EPH136 on DNA and RNA synthesis. In Burkitt’s lymphoma cells the IC50 for inhibition of cell proliferation is 36 nM, that for inhibition of DNA or RNA synthesis is >5 μM (Fig. 4). Incorporation of cytidine into DNA as a measure for deoxycytidine synthesis from cytidine (a measure for in vivo inhibition of ribonucleotide reductase) by EPH136 was affected more than that of DNA and RNA synthesis (Fig. 4). Hydroxyurea, an antitumor drug and inhibitor of ribonucleotide reductase, inhibits this enzyme with an IC50 of 37.2 μM and the proliferation of Burkitt’s lymphoma cells with an IC50 of 140 μM. For hydroxyurea the ratio of IC50 of ribonucleotide reductase/IC50 cell proliferation is 0.26 [8]. For EPH136 the IC50 for cytidine incorporation into DNA was found to be 216 nM, that for cell proliferation 36 nM, and thus this ratio was 6. In addition to the results of the cell cycle experiments (Fig. 3B), this is an indication that ribonucleotide reductase is not the main target of EPH136.

Inhibition of DNA synthesis, RNA synthesis and ribonucleotide reductase by EPH136. Incorporation of labeled cytidine, thymidine and uridine into DNA and RNA, respectively, was determined in presence of the indicated concentrations of EPH136. The mean (±SD) of two independent experiments, in which two samples were taken within each experiment, is shown
The COMPARE algorithm was employed to identify the mechanism of action of EPH136 by comparison with the pattern of known compounds. A correlation coefficient of 0.55–0.6 is considered to be the lowest correlation that suggests a relationship of a seed compound with others [20]. The COMPARE analysis of the effects of EPH136 as seed has shown that the highest correlations with standard agents with known mechanisms of action are 0.425 (OH-2-formylpyridine TSC, DNA antimetabolite) and 0.39 (Brequinar, DNA/RNA antimetabolite). Because these correlation coefficients with the standard agents are well below 0.55, the mechanism of action of EPH136 must be considered to be different from those of the 175 standard antitumor drugs. Therefore, the novel compound and other related compounds [6–9] exhibit a new and unknown mechanism of action and we tried to find clues about the target.
Bioinformatic approach
In order to find out which genes are involved in the inhibition of cell proliferation by EPH136, the multivariate statistical procedure PLS included in the SIMCA package was used. With this bioinformatic approach the expression of 9,799 variables in each of the untreated cell lines was related to the response (log GI50) of each cell line following treatment with EPH136. The present work considers an X matrix, where 56 cell lines (i.e. objects) may be represented as characterized by a multivariate biological fingerprint, given by the gene expression profiles or by molecular targets (the “descriptor” variables) and a dependent variable (y) expressed as log GI50.
The PLS model included 56 cell lines (objects) with different tissue origins (lung, colon, breast, ovarian, leukaemia, renal, melanoma, prostate, CNS) taken from the original NCI 60 cell line panel. Two breast cancer cell lines (MDA-MB435, MDA-N) and one melanoma line (LOX IMVI) were excluded on the basis of previously reported considerations [13, 14, 21], and one renal cancer cell line (TK-10) was excluded due to the lack of the dependent variable (y).
The PLS procedure extracted four significant components explaining 98.5% y variance, with a satisfactory predicting ability (Q 2 = 0.457). The VIP values of the PLS model represent a condensed summary of the importance of the X variables and therefore can be considered as proper statistical parameter to select the main gene expression targets involved in cancer chemotherapy for the selected drug. The coefficients provide a useful indication on the correlation between molecular target level and biological response to the drug. The top 60 most important genes as classified by this procedure are shown in Table 1, together with their VIP values. It is perhaps worth mentioning here that approximately one half (29 out of 60) of the top genes in Table 1 are either unknown or expressed sequence tags (ESTs). This list of genes provides a bioinformatics clue for future investigations aimed at a better genome-based understanding of EPH136 mechanism of action. The following considerations will therefore regard only the 31 known genes in Table 1. The genes from Table 1 were grouped with Panther software (http://www.pantherdb.org/genes/batchIdSearch.jsp) according to their involvement in biological processes. The following groups were found (proteins are cited more than once if they belong to more than one group):
Nucleoside, nucleotide, nucleic acid metabolism and binding: uracil-DNA glycosylase, casein kinase 1 alpha 1, HGPRT, nuclear receptor subfamily 2, ATP synthase F0 subunit, ELAV.
Developmental processes: myosin, heavy chain, casein kinase 1 alpha 1, cadherin 8, nuclear receptor subfamily 2, VEGF, contactin 2, ELAV.
Protein modification and metabolism: protein kinase C substrate 80 kDa, casein kinase 1 alpha 1, coagulation factor VIII, mannosidase II alpha, ser/thre kinase 17a, casein kinase 1 alpha 1, PI3-kinase regulatory subunit 85 K, SMTA3 protein, ERK3, PTEN, microsomal HSP 70, crystallin alpha B.
Gene expression by microarray
As another approach to obtain clues about the mechanism of action of EPH136 we used DNA microarray technology. Following treatment of HT-29 colon carcinoma cells with 80 nM EPH136 (approximately two-fold IC50) for 24 h we measured the expression of ~5,000 known genes in comparison with untreated controls. Three genes were found to be down-modulated: v-erbA (viral oncogene, nuclear receptor), the SW1/SNF-related, matrix-associated, actin-dependent regulators of chromatin (SMARC), and the neutral amino acid transporter. The genes which were up-regulated more than two-fold are shown in Table 2. As shown below, according to Panther software the overexpressed genes belong to similar groups as those which were calculated to be important for the antiproliferative activity of EPH136: Nucleoside, nucleotide, nucleic acid metabolism and binding: eIF-4E, microsomal HSP70, heterochromatin protein p25, cartilage-specific homeoprotein, melanocyte-specific gene 1, ras-related small GTP-binding protein, TAR-protein, bone morphogenetic protein, Hin3/HIV chimeric promoter, transcription regulator ISGF3, ATP-binding protein RNF31, X-linked helicase, transcriptional repressor ZEM1 B3, GAP SH3-binding protein, pp21 transcriptional elongation factor, heterochromatin protein gamma, CREB5, splicing factor SRp30c, myosin light chain, acid finger protein ZNF 173, RLIP76, core-binding factor beta, TGF-beta superfamily protein, zinc finger protein 24, 8-oxoguanine DNA glycosylase hOGG1, ribophorin, STAT5B, catenin beta, protoporphyrinogen oxidase, RhoE.
Developmental processes: myosin light chain, germline oligomeric matrix protein, cartilage-specific homeoprotein 1, TAR RNA binding protein, bone-morphogenetic protein 2, ribosomal protein S6 kinase, STAT5B, cadherin 11.
Protein modification and metabolism: PI3-kinase, eIF-4E, microsomal HSP 70 protein, activin A receptor, type II, metallopeptidase 1, TAR protein, serine peptidase inhibitor, chymotrypsinogen B1, cystatin B, ribosomal protein S6 kinase, SMTA3 protein, ERK3, protein tyrosine phosphatase, PTEN, acid finger protein ZNF173, autocrine motility factor receptor, TRAF-interacting protein, CLK2 CDC-like kinase, O-linked GlcNAc transferase, ribophorin I, crystallin alpha B.
The results shown in Table 2 were obtained following treatment with a concentration corresponding to the two-fold IC50 for 24 h. To confirm these results, this experiment was also performed with a two-fold IC50 concentration of EPH136 for 12 h and an IC50 concentration for 12 and 24 h. In these experiments the results shown in Table 2 were confirmed, although the highest levels of gene expression were obtained with a two-fold IC50 treatment for 24 h (shown in Table 1).
The genes found by the bioinformatic and the gene array approaches, can be grouped into the same classes of genes although the genes in both approaches are different. Exceptions are PI3 kinase and eIF-4E. These are the genes with the highest rankings in both approaches (VIP value 2.07 in Table 1, gene 2 in Table 2; VIP value 1.77, not shown in Table 1, gene 3 in Table 2, respectively). Experiments demonstrated that EPH136 did not inhibit PI3-kinase or other members of the PI3-kinase pathway (data not shown) indicating that it is not the target. It has been reported that PI3-kinase and eIF-4E are involved in the response of cells to oxidative stress [22,23]. In Tables 1 and 2 a series of transcription factors, kinases and phosphatases can be found. Many of these proteins are regulated by oxidation/reduction [24–28]. Therefore, we investigated whether EPH136 can induce the formation of ROS.
Formation of ROS
As shown in Fig. 5, the compound led to a dose-dependent increase of luminol-induced fluorescence, indicating production of ROS. It was shown previously that also thalidomide analogs exhibited antitumor activity via induction of ROS and this led to dissipation of the mitochondrial membrane potential [29]. Therefore, we tested the effects of EPH136 on the mitochondrial membrane potential. As shown in Table 3 by the fluorescent dye JC-1, EPH136 indeed induced dissipation of the mitochondrial membrane potential 24 h after treatment. 14 h after treatment with EPH136, disruption of the mitochondrial network and swelling of mitochondria was observed in the microscope (Fig. 6).

Luminescence following treatment with EPH136. Luminescence as measure for ROS was detected as described in “Materials and Methods”. The means (±SD) of three independent experiments, in which two samples were taken, are indicated. (PBS Phosphate buffered saline)

Real time live confocal imaging of mitochondria. Red is TMRM showing mitochondria with an active potential, green is WGA showing cell morphology. The frames show areas typical for untreated and treated cells. a Untreated Burkitt’s lymphoma CA46 control cells. b Burkitt’s lymphoma CA46 cells treated with 100 nM EPH136 for 14 h
Prevention of the antiproliferative activity by antioxidants
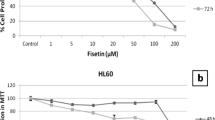
If ROS is important for the antiproliferative activity of EPH136, its removal should prevent antiproliferative activity. Indeed, the antioxidant NAC prevented EPH136 from exercising inhibition of cell proliferation (Fig. 7A). These data illustrates that radicals are involved in the antiproliferative activity of the novel compound. A2780CP ovarian carcinoma cells are resistant to cis-platin due to elevated levels of glutathione if compared with parental A2780 cells [30]. A2780CP cells exhibited also resistance to EPH136 (Fig. 7B), indicating that the antioxidant activity of glutathione reduces the activity of EPH136.

A Inhibition of cell proliferation of Burkitt’s lymphoma cells by EPH136 and adriamycin in the presence and absence of NAC as described in “Materials and Methods”. The means (±SD) of two independent experiments, in which two samples were taken within each experiment, are shown. B Antiproliferative activity of EPH136 in parental A2780 and A2780CP cells that exhibit elevated levels of glutathione. The means (± SD) of six independent experiments, in which two samples were taken, are indicated
Discussion
Bicyclic hydrazones represent a new class of experimental antitumor agents. The mechanism of action of these compounds was not known [6–9]. EPH136 inhibited ribonucleotide reductase (Fig. 4), however the ratio of IC50 of ribonucleotide reductase/IC50 cell proliferation indicated that it is not the main mechanism of action. A COMPARE analysis also showed no correlation with ribonucleotide reductase inhibitors. We screened the effects of EPH136 on cell cycle kinases, EGFR, ErbB2, PKB/Akt, IGFR1 and FLT-4 kinase activities, as well as Na+/K+-ATPase. No effects on these parameters were found (data not shown). Instead of further random testing in different systems, we employed novel approaches for identifying the target of EPH136. In a bioinformatic approach, using the multivariate statistical PLS procedure, we identified genes important for the inhibition of cell proliferation by EPH136. The principle is based on the correlation of log GI50 values with the expression levels of 9,799 variables (representing approximately 8,000 genes) in the NCI panel of 56 cell lines. Interestingly, only 679 of the original 9,799 variables considered in the PLS analysis exhibited VIP values above 1.5. The top 60 genes found to be important for the antiproliferative activity of EPH136 (Table 1) are involved in nucleoside, nucleotide, nucleic acid metabolism and binding, developmental processes, protein modification and metabolism.
Following treatment of HT-29 cells EPH136 for 24 h, in a microarray the genes found to be overexpressed more than two-fold, belonged to the same groups of genes as those found by the bioinformatic approach. If the bioinformatics analysis is extended to include 679 genes with VIP coefficients in the PLS model above 1.50 (threshold well above that considered to be important on a safe statistical basis) [15], it is possible to identify the following nine genes that are highly induced by EPH136 treatment (fold-induction above 2) and statistically significant in influencing the EPH136 response (numbers in parentheses indicate rank position according to fold-induction in Table 2 and according to the VIP value in Table 1, respectively): PI3-kinase (no. 2 and 31), eIF-4E (no. 3 and 160), fetal brain (239FB) protein (no. 14 and 172), TAR protein (no. 19 and 597), Coch-5B2 (no. 28 and 276), splicing factor SRp30c (no. 38 and 360), protein tyrosine phosphatase (no. 42 and 649), crystallin alpha B (no. 72 and 553) and RhoE (no. 73 and 434).
The above correlations may be regarded as satisfactory, as discrepancies between Tables 1 and 2 are not unexpected due to the fact that the bioinformatic approach picks important genes for EPH136 treatment of 56 cell lines from different tissue histotypes, while Table 2 reports experimental data for only one colon cancer cell line (HT-29). Therefore it is not surprising that top genes selected on VIP values mediated from 56 cells, which might derive from a high contribution of other cells and a low contribution of HT-29, are not in Table 2. Furthermore, the basic meaning of the two approaches is different. Experiments that evaluate the effect of EPH136 on global gene expression profile lead only to genes that are up- or down-regulated following the drug treatment, while the bioinformatic approach tends to select genes the basal expression of which is able to influence the response to the drug.
As stated in the paragraph above, the genes with the highest rankings in both approaches are PI3-kinase and eIF-4E. It has to be emphasized that with the first approach the regulatory and with the second approach the catalytic subunits of PI3-kinase was found. PI3-kinase is an attractive target for cancer chemotherapy. This oncogene is involved in tumor development, apoptosis, and also related to the resistance to 5-FU, adriamycin and cyclophosphamide by activation of the PKB/akt survival pathway [17, 31]. It is well known that PI3-kinase and eIF-4E are functionally strictly related. eIF-4E binds to the 5′-cap structure of eukaryotic mRNA and plays a central role in the control of cell proliferation [32]. A complex is formed by interactions between eIF-4E and other factors such as eIF-4F and eIF-4G, which binds to the 5′ mRNA cap structure and permits the recruitment of the eukaryotic ribosome. eIF-4E activity is blocked by interaction with 4E-binding proteins (4E-BPs) and this interaction is regulated by phosphorylation: hypo-phosphorylated 4E-BPs strongly bind eIF-4E while hyper-phosphorylated 4E-BPs release eIF-4E. The phosphorylation of 4E-binding protein is regulated by PI3-kinase, which represents an important link between PI3-kinase activity and eIF-4E [24, 25]. However, our experiments showed that EPH136 did not inhibit PI3-kinase or its pathway (data not shown). It has been reported that PI3-kinase and eIF-4E are involved in the response of the cell to oxidative stress [22, 23]. A series of transcription factors, kinases and phosphatases are also regulated by oxidation/reduction [26–28]. It was shown that PI3-kinase mediated mitochondrial ROS production [33], and ROS stimulated PI3-kinase [34]. Therefore, the appearance of PI-3 kinase and eIF-4E in both approaches can be explained. As shown in Fig. 5, EPH136 exhibited a dose-dependent increase in luminol-dependent luminescence, indicating an increase in ROS. The radical scavenger NAC prevented the antiproliferative activity of EPH136 (Fig. 7A) and elevated levels of the antioxidant glutathione reduced its antiproliferative activity (Fig. 7B). These results illustrate that radical formation is indeed involved in the mechanism of action of the compound. It has been shown that adriamycin, a topoisomerase II inhibitor, also generates ROS [35]. As shown in Fig. 7A, NAC did not alter the antiproliferative activity of adriamycin. This seems to be an indication that at least in Burkitt’s lymphoma cells ROS are not involved in the antiproliferative activity of adriamycin. If radical formation is the main mechanism of EPH136 and most likely also that of other bicyclic hydrazones, it would be an intriguing mechanism for antitumor activity. Malignant cells in general are more active than normal cells, are under intrinsic oxidative stress, and thus are more vulnerable to damage by ROS-generating agents [36]. ROS have been causatively implicated in a variety of cell death pathways including non-apoptotic death [30, 37]. This may explain the low level of apoptosis after treatment with EPH136 (Fig. 3A, Table 3). Also thalidomide analogs exhibiting selective leukemic-cell killing due to ROS generation induce necrotic-programmed cell death [30]. Many tumor cells exhibit relatively low levels of superoxide dismutase, which leads to high levels of oxygen radicals [38]. ROS have the ability to stimulate ceramide synthesis leading to cell death [39]. Therefore, it was proposed previously that one way of treating cancer might be the design of drugs to target enzymes that increase ROS [36]. One such drug is 2-methoxyoestradiol, an inhibitor of superoxide dismutase which causes accumulation of ROS and kills selectively cancer cells [40]. Aplidin, a peptide generating ROS also exhibits antitumor activity [41]. Verrax et al. used a combination of vitamin K3 and vitamin C to develop an oxidant insult [42]. EPH136 seems to be another compound with this kind of mechanism of action. In order to explain the exact mechanism, detailed investigations into the kind of radicals and their generation are essential.
References
Mauro MJ, O’Dwyer M, Heinrich MC, Druker BJ (2002) STI571: a paradigm of new agents for cancer therapeutics. J Clin Oncol 20(1):325–334 doi:10.1200/JCO.20.1.325
Folkman J (2001) Angiogenesis-dependent diseases. Semin Oncol 28(6):536–542 doi:10.1016/S0093-7754(01)90021-1
Lothstein L, Israel M, Sweatman TW (2000) Anthracycline drug targeting: cytoplasmic versus nuclear—a fork in the road. Drug Resist Updat 14:169–177
Jansen B, Wacheck V, Heere-Ress E, Schlagbauer-Wadl H, Hoeller C, Lucas T et al (2000) Chemosensitisation of malignant melanoma by BCL2 antisense therapy. Lancet 356(9243):1728–1733 doi:10.1016/S0140-6736(00)03207-4
Yuen AR, Halsey J, Lum B, Fisher G, Holmlund JT, Geary R et al (1999) Phase I study of an antisense oligonucleotide to protein kinase C-α, (ISIS 3521/CGP64128A) in patients with cancer. Clin Cancer Res 5(11):3357–3363
Easmon J, Heinisch G, Hofmann J, Langer T, Grunicke HH, Fink J et al (1997) Thiazoyl and benzothiazoyl hydrazones derived from α-(N)-acetylpyridines and diazines: synthesis, antiproliferative activity, and CoMFA studies. Eur J Med Chem 32:397–408 doi:10.1016/S0223-5234(97)81677-7
Easmon J, Heinisch G, Pürstinger G, Langer T, Österreicher JK, Grunicke HH et al (1997) Azinyl and diazinyl hydrazones derived from aryl N-heteroaryl ketones: synthesis and antiproliferative activity. J Med Chem 40(26):4420–4425 doi:10.1021/jm970255w
Easmon J, Puerstinger G, Roth T, Fiebig HH, Jenny M, Jaeger W et al (2001) Benzoxazolyl and 2-benzimidazolyl hydrazones derived from 2-acetylpyridine: a novel class of antitumor agents. Int J Cancer 94(1):89–96 doi:10.1002/ijc.1427
Easmon J, Puerstinger G, Heinisch G, Hofmann J (2006) Synthesis, structure-activity relationships, and antitumor studies of 2-benzoxazolyl hydrazones derived from alpha-(N)-acyl heteroaromatics. J Med Chem 49(21):6343–6350 doi:10.1021/jm060232u
Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L et al (1989) Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J Natl Cancer Inst 81(14):1088–1092 doi:10.1093/jnci/81.14.1088
Weinstein JN, Kohn KW, Grever MR, Viswanadhan VN, Rubinstein LV, Monks AP et al (1992) Neural computing in cancer drug development: predicting mechanism of action. Science 258(5081):447–451 doi:10.1126/science.1411538
Musumarra G, Condorelli DF, Costa AS, Fichera M (2001) A multivariate insight into the in vitro antitumour screen database of the National Cancer Institute: classification of compounds, similarities among cell lines and the influence of molecular targets. J Comput Aided Mol Des 15(3):219–234 doi:10.1023/A:1008171426412
Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L et al (2000) A gene expression database for the molecular pharmacology of cancer. Nat Genet 24(3):236–244 doi:10.1038/73439
Musumarra G, Condorelli DF, Scire S, Costa AS (2001) Shortcuts in genome-scale cancer pharmacology research from multivariate analysis of the National Cancer Institute gene expression database. Biochem Pharmacol 62(5):547–553 doi:10.1016/S0006-2952(01)00711-0
Wold S (1998) PLS in Chemistry. In: Schleyer PVR (ed) The encyclopedia of computational chemistry. Wiley, Chichester, pp 2006–2020
Barresi V, Fortuna CG, Garozzo R, Musumarra G, Scirè S, Condorelli DF (2006) Identification of genes involved in the sensitivity to antitumour drug 17-allylamino,17-demethoxygeldanamycin (17AAG). Mol Biosyst 2(5):231–239 doi:10.1039/b518093g
Zembutsu H, Ohnishi Y, Tsunoda T, Furukawa Y, Katagiri T, Ueyama Y et al (2002) Genome-wide cDNA microarray screening to correlate gene expression profiles with sensitivity of 85 human cancer xenografts to anticancer drugs. Cancer Res 62(2):518–527
Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139(2):271–279 doi:10.1016/0022-1759(91)90198-O
Weiss G, Fuchs D, Hausen A, Reibnegger G, Werner ER, Werner-Felmayer G et al (1993) Neopterin modulates toxicity mediated by reactive oxygen and chloride species. FEBS Lett 321(1):89–92 doi:10.1016/0014-5793(93)80627-7
Weinstein JN, Myers TG, O’Connor PM, Friend SH, Fornace AJ Jr, Kohn KW et al (1997) An information-intensive approach to the molecular pharmacology of cancer. Science 275(5298):343–349 doi:10.1126/science.275.5298.343
Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P et al (2000) Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 24(3):227–235 doi:10.1038/73432
Rao GN (2000) Oxidant stress stimulates phosphorylation of eIF4E without an effect on global protein synthesis in smooth muscle cells. Lack of evidence for a role of H202 in angiotensin II-induced hypertrophy. J Biol Chem 275(22):16993–16999 doi:10.1074/jbc.275.22.16993
Kang KW, Lee SJ, Park JW, Kim SG (2002) Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol Pharmacol 62(5):1001–1010 doi:10.1124/mol.62.5.1001
Fleurent M, Gingras AC, Sonenberg N, Meloche S (1997) Angiotensin II stimulates phosphorylation of the translational repressor 4E-binding protein 1 by a mitogen-activated protein kinase-independent mechanism. J Biol Chem 272(7):4006–4012 doi:10.1074/jbc.272.7.4006
Martin KA, Blenis J (2002) Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv Cancer Res 86:1–39
Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T (1995) Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270(5234):296–299 doi:10.1126/science.270.5234.296
Ryter SW, Tyrrell RM (1998) Singlet molecular oxygen ((1)O2): a possible effector of eukaryotic gene expression. Free Radic Biol Med 24(9):1520–1534 doi:10.1016/S0891-5849(97)00461-9
Zafarullah M, Li WQ, Sylvester J, Ahmad M (2003) Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci 60(1):6–20 doi:10.1007/s000180300001
Ge Y, Montano I, Rustici G, Freebern WJ, Haggerty CM, Cui W et al (2006) Selective leukemic-cell killing by a novel functional class of thalidomide analogs. Blood 108(13):4126–4135 doi:10.1182/blood-2006-04-017046
Abe T, Gotoh S, Higashi K (1999) Attenuation by glutathione of hsp72 gene expression induced by cadmium in cisplatin-resistant human ovarian cancer cells. Biochem Pharmacol 58(1):69–76 doi:10.1016/S0006-2952(99)00049-0
Roymans D, Slegers H (2001) Phosphatidylinositol 3-kinases in tumor progression. Eur J Biochem 268(3):487–498 doi:10.1046/j.1432-1327.2001.01936.x
Fraser CS, Pain VM, Morley SJ (1999) Cellular stress in xenopus kidney cells enhances the phosphorylation of eukaryotic translation initiation factor (eIF)4E and the association of eIF4F with poly(A)-binding protein. Biochem J 342(3):5119–5126 doi:10.1042/0264-6021:3420519
Park HS, Lee SH, Park D, Lee JS, Ryu SH, Lee WJ et al (2004) Sequential activation of phosphatidylinositol 3-kinase, beta Pix, Rac1, and Nox1 in growth factor-induced production of H2O2. Mol Cell Biol 24(10):4384–4394 doi:10.1128/MCB.24.10.4384-4394.2004
Lee SB, Bae IH, Bae YS, Um HD (2006) Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem 281(47):36228–36235 doi:10.1074/jbc.M606702200
Müller I, Niethammer D, Bruchelt G (1998) Anthracycline-derived chemotherapeutics in apoptosis and free radical cytotoxicity. Int J Mol Med 1(2):491–494
Cleveland JL, Kastan M (2000) A radical approach to treatment. Nature 407(6802):309–311 doi:10.1038/35030277
Shen HM, Liu ZG (2006) JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med 40(6):928–939 doi:10.1016/j.freeradbiomed.2005.10.056
Das UN (2002) A radical approach to cancer. Med Sci Monit 8(6):RA79–RA92
Huwiler A, Boddinghaus B, Pautz A, Dorsch S, Franzen R, Briner VA et al (2001) Superoxide potently induces ceramide formation in glomerular endothelial cells. Biochem Biophys Res Commun 284(2):404–410 doi:10.1006/bbrc.2001.4941
Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W (2000) Superoxide dismutase as a target for the selective killing of cancer cells. Nature 407(6802):390–395 doi:10.1038/35030140
Garcia-Fernandez LF, Losada A, Alcaide V, Alvarez AM, Cuadrado A, Gonzalez L et al (2002) Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C delta. Oncogene 21(49):7533–7544 doi:10.1038/sj.onc.1205972
Verrax J, Taper H, Calderon BP (2008) Targeting cancer cells by an oxidant-based therapy. Curr Mol Pharmacol 1:80–92
Acknowledgments
The work was funded by grants P09879-MED and P12384-MOB from the Austrian Science Fund. We thank Dr. Angelika Burger, Department of Pharmacology and Experimental Therapeutics, Marlene and Stewart Greenebaum Cancer Center, University of Maryland School of Medicine, Baltimore, MD 21201, USA, for both A2780 cell lines. We also want to thank the National Cancer Institute, Bethesda, MD, for screening the compound in the panel of cell lines.
Conflict of interest statement
Austria Wirtschaftsservice, a governmental organization which files patents for innovations of Austrian universities, obtained a patent on EPH136 and related compounds. The authors J. Hofmann, J. Easmon, G. Puerstinger and G. Heinisch are coassignees of the patent.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hofmann, J., Easmon, J., Puerstinger, G. et al. N-benzoxazol-2-yl-N′-1-(isoquinolin-3-yl-ethylidene)-hydrazine, a novel compound with antitumor activity, induces radicals and dissipation of mitochondrial membrane potential. Invest New Drugs 27, 189–202 (2009). https://doi.org/10.1007/s10637-008-9156-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-008-9156-x