
Abstract
NME4, also designated nm23-H4 or NDPK-D, has been known for years for its well-established roles in the synthesis of nucleoside triphosphates, though; little has been known regarding the differential metabolites involved as well as the biological roles NME4 plays in proliferation and invasion of esophageal squamous cell carcinoma (ESCC) cells. To understand the biological roles of NME4 in ESCC cells, lentiviral-based short hairpin RNA interference (shRNA) vectors were constructed and used to stably knock down NME4. Then, the proliferative and invasive variations were assessed using MTT, Colony formation and Transwell assays. To understand the metabolites involved after silencing of NME4 in ESCC cells, widely targeted metabolomic screening was taken. It was discovered that silencing of NME4 can profoundly suppress the proliferation and invasion in ESCC cells in vitro. Metabolically, a total of 11 differential metabolites were screened. KEGG analyses revealed that Tryptophan, Riboflavin, Purine, Nicotinate, lysine degradation, and Linoleic acid metabolism were also involved in addition to the well-established nucleotides metabolism. Some of these differential metabolites, say, 2-Picolinic Acid, Nicotinic Acid and Pipecolinic Acid were suggested to be associated with tumor immunomodulation. The data we described here support the idea that metabolisms occurred in mitochondrial was closely related to tumor immunity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nucleoside diphosphate kinases (NDPK) are nucleotide metabolism enzymes encoded by NME, also termed NM23 genes [1]. So far, as many as 10 genes have been identified to be part of the NDPK/NME family [2]. Among the NME genes, NME4 has been little described and defined in cancers, not to mention in ESCC, with the exception of only one recent study [3] performed surrounding NME4 in non-small cell lung cancer; which stands in stark contrast to the NME1, NME2 or NME3, the paralogs of NME4 in NME gene family, which have been extensively investigated in cancers [4, 5]. On the other hand, although the well-established roles of NME4 in nucleotide metabolism [6] have been known for many years, the metabolites mediated by NME4 in cancer cells remain far less understood. In the present study, we seek to understand the biological roles played by NME4 in ESCC cells and to work out the differential metabolites mediated by NME4, with the aid of widely targeted metabolomics.
In our study, it was discovered that silencing of NME4 was shown to slow the proliferation and invasion of ESCC cells in vitro, indicative of the tumor-promoting roles in ESCC cells. With the help of widely targeted metabolomic screening, a total of 11 differential metabolites were identified, specifically, with 7 being upregulated and 4 downregulated. These differential metabolites identified comprise Inosine, Hypoxanthine-9-β-d-Arabinofuranoside, 15-oxoETE [15-oxo-5Z,8Z,11Z,13E-eicosatetraenoic acid], 2-Picolinic Acid, B-Nicotinamide Mononucleotide, Nicotinic Acid, 6-Methylnicotinamide, Lumichrome, Pantothenol, Pipecolinic Acid and 12,13-EpOME [(±)12(13)epoxy-9Z-octadecenoic acid]. Some of these metabolites, for example, 2-Picolinic Acid, Nicotinic Acid and Pipecolinic Acid were suggested to be associated with tumor immunomodulation. Our data shown here firstly defined the oncogenic trait of NME4 in ESCC cells, and our data support the notion that metabolisms occurred in mitochondrial was related to tumor immunity.
Materials and methods
Cell culture and lentivirus-mediated shRNA transfection
ESCC cell lines TE-1, Eca109 and EC9706 cell lines were commercially from Wuhan University (Wuhan, China). All cells were cultured by RPMI 1640 medium (Beijing Solarbio Science and Technology Co., Ltd, Beijing, China) containing 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) and 100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C in humidified 5% CO2 incubator. Lentiviruses carrying green fluorescent protein along with scrambled (shRNA-control), or shRNA-NME4 (shRNA-NME4) were purchased from Shanghai GenePharma Co. Ltd. (Shanghai, China). For infection, TE-1 and EC9706 cells were incubated with lentiviruses [multiplicity of infection (MOI) of 15] using polybrene (5 μg/mL) and enhanced infection solution following the manufacturer’s protocol. After 72 h, all fluorescent cells were sorted via flow cytometry. The sorted cells were treated with hygromycin (800 μg/mL). The shRNA-NME4 target sequence was 5′-CCGGCATGATTGGACACACCGACTCCTCGAGGAGTCGGTGTGTCCAATCATGTTTTT-3′.
RNA extraction and qRT-PCR assay
Total RNA were extracted from ESCC cells with Trizol (Invitrogen, Carlsbad, CA, USA), followed by reverse transcription into cDNA using the PrimeScript RT reagent Kit (Catalog Number RR047A; TaKaRa, Dalian, China) following the manufacturer’s protocol. cDNA was amplified and was detected by an ABI 7500 fast system (Applied Biosystems, CA, USA). GAPDH was used as internal control for NME4 relative expression. The primer sequences used were as follows: GAPDH, F-5′-GGAGCGAGATCCCTCCAAAAT-3′; R-5′-GGCTGTTGTCATACTTCTCATGG-3′; NME4, F: 5′-CAACTACCTCCGTCAGCAAGAACC-3′; R: 5′-GTTGGTGGCTCAAACTCCTCTGG-3′. \(2^{{ - \Delta \Delta c_{{\text{t}}} }}\) method was taken to calculate the relative expression of NME4. The annealing temperature was set at 55 °C throughout. The qRT-PCR reactions were performed with ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA).
Western-blot
Total proteins were extracted with Radio-Immunoprecipitation Assay (RIPA) lysis buffer (CWbiotech, Beijing, China) containing the protease inhibitor cocktails (Catalog Number 4693116001; cOmplete; Roche, Switzerland), and the concentration was determined by BCA kit (Solarbio, Beijing, China). The proteins were separated by running 12% SDS-PAGE gel and transblotted to PVDF membranes. Then, membranes were blocked with 5% skimmed milk for 2 h and incubated with the primary antibody anti-NME4 (1:1000; Catalog Number GTX121931, GeneTex, CA, USA), anti-GAPDH (1:1000; Catalog Number TA-08; ZSGB-BIO, Beijing, China) overnight at 4 °C and with secondary antibodies (horseradish-conjugated goat anti-rabbit) for 2 h. The bands were visualized using GelDox XR System (Bio-Rad, CA, USA). Quantity One software (Bio-Rad, CA, USA) was used to quantify the variation of band grey.
MTT
TE-1 and EC9706 cells were seeded at a density of 4 × 103/well in a 96-well plate and incubated overnight. At 0, 24, 48, 72, and 96 h, 20 μL (5 g/L) of MTT (3-[4, 5-dimethyl-2-thiazolyl]-2, 5-diphenyl-2-H-tetrazolium bromide) was added to the designated wells. After a 4 h incubation, the MTT formazan precipitate was dissolved in dimethylsulfoxide (DMSO, 150 μL/well, Sigma-Aldrich, St. Louis, MO, USA) in a shaker before reading the absorbance at 490 nm using a 96-well plate reader (Bio-Rad, Winooski, VT, USA).
Colony formation assay
For colony formation assay, cells were seeded evenly in 6-well plates (4 × 102 cells/well) and continuously cultured for 14 days. Then the cells were fixed with methanol for 10 min, stained with 1% crystal violet staining solution for 10 min.
Transwell assay
The invasion assays were performed using a 24-well Transwell chamber with a polycarbonate membrane with a pore size of 8 μm (Corning, NY, USA). The membrane was coated with 60 μL of a 1:5 mixture of matrigel (BD Sciences, San Jose, CA, USA) and serum-free RPMI 1640 medium to form a matrix barrier. After the matrigel was allowed to solidify at 37 °C for 2 h, TE-1 and EC9706 (1 × 105 cells/mL) were added to the upper chamber without serum; the lower chamber was filled with 0.6 mL of medium with 10% FBS. After incubation at 37 °C for 24 h, invaded cells were then rinsed with PBS and stained with crystal violet. The cells remaining at the top of the polycarbonate membrane were removed using cotton swab. The cell numbers in four random fields were manually counted under magnification of 200-fold.
Apoptosis assay
The apoptosis assay was performed with flow cytometry using the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) following the manufacturer’s instruction. In brief, Cells were washed twice with cold PBS and resuspended in 400 mL with 1 × binding buffer at a concentration of 1 × 106 cells/mL. Cells were then mixed with 5 mL of the Annexin V-FITC solution and 2 mL of propidium iodide, followed by incubation for 15 min at 4 °C in the dark, finally have these cells analyzed by flow cytometry.
Cell lysate sample
Cell lysate samples from TE-1 and EC9706 cell lines were thawed on ice, then added 1 mL pre-cooled extractants (70% methanol aqueous solution), and whirl for 1 min. Freeze the mixture for 3 min in liquid nitrogen after remove ice for 3 min, it will be whirled for 2 min, circulate this at 3 times. Centrifuge the mixture again with 12,000 r/min at 4 °C for 10 min. Finally take the supernatant into the sample bottle for LC–MS/MS analysis.
HPLC conditions
The sample extracts were analyzed using an LC–ESI–MS/MS system (UPLC, Shim-pack UFLC Shimadzu CBM A system, https://www.shimadzu.com/; MS, QTRAP® System, https://sciex.com/). The analytical conditions were as follows, UPLC: column, Waters Acquity UPLC HSS T3 C18 (1.8 µm, 2.1 mm * 100 mm); column temperature, 40 °C; flow rate, 0.4 mL/min; injection volume, 2 μL; solvent system, water (0.04% acetic acid): acetonitrile (0.04% acetic acid); gradient program, 95:5 V/V at 0 min, 5:95 V/V at 11.0 min, 5:95 V/V at 12.0 min, 95:5 V/V at 12.1 min, 95:5 V/V at 14.0 min.
ESI–QTRAP-MS/MS
LIT and triple quadrupole (QQQ) scans were acquired on a triple quadrupole-linear ion trap mass spectrometer (QTRAP), QTRAP® LC–MS/MS System, equipped with an ESI Turbo Ion-Spray interface, operating in positive and negative ion mode and controlled by Analyst 1.6.3 software (Sciex). The ESI source operation parameters were as follows: source temperature 500 °C; ion spray voltage (IS) 5500 V (positive), − 4500 V (negative); ion source gas I (GSI), gas II (GSII), curtain gas (CUR) were set at 55, 60, and 25.0 psi, respectively; the collision gas (CAD) was high. Instrument tuning and mass calibration were performed with 10 and 100 μmol/L polypropylene glycol solutions in QQQ and LIT modes, respectively. A specific set of MRM transitions were monitored for each period according to the metabolites eluted within this period.
Statistical analysis
All experiments were carried out independently three times, unless otherwise stated. GraphPad Prism 8.0 software (GraphPad Software, Inc., La Jolla, CA, USA) was used to generate the plots presented and to statistically analyze the difference. The data were expressed as mean ± standard deviation. A two-tailed, unpaired Student’s T-test was used for statistical analysis. Two-tailed, unpaired T-test with Welch’s correction was used to analyze the difference between two groups. Metabolite data were log2-transformed for statistical analysis to improve normality and normalized. To classify the differential metabolites screened, the 346 metabolites data have been used for cluster analysis by R (www.r-project.org/). The color scale indicates the intensity of the metabolites (log2-transformed). Unsupervised principal content analysis was conducted by SPSS (Version 18.0, SPSS, Inc., Chicago, IL, USA). Differences in the metabolites between NME4 stably knock-down and control were determined using Welch’s T-test (P < 0.01). Difference was considered to be statistically significant when P < 0.05.
Results
NME4 promotes proliferation and invasion of ESCC cells in vitro
To start with, baseline expression of endogenous NME4 was detected on transcriptional level using qRT-PCR technique in the three different ESCC cell lines we enrolled in our experimental setting. In terms of basal NME4 mRNA expression, TE-1 and EC9706 whose NME4 were shown to be remarkably higher than that of ECa109 cell line (Fig. 1a). Therefore, TE-1 and EC9706 were picked up in the experiment that follows. To understand the biological roles NME4 mediates in the growth and motility of ESCC cells, lentiviral-based shRNA vectors targeting against NME4 were constructed and transfected into TE-1 and EC9706 cells, respectively. Knock-down efficiency was evaluated using western-blot after sorting of cells. It was exhibited that NME4 were significantly silenced in TE-1 and EC9706 cells (Fig. 1b, c). Based on the interferential effect displayed, the roles of NME4 mediate in proliferation and invasion were appraised by MTT, colony formation and Transwell assays. It was exhibited that, compared with control group, proliferative abilities of ESCC cells were markedly slowed down after NME4 were stably knocked down (Fig. 1d); which were corroborated by colony formation assay undertaken in parallel (Fig. 1e). Meanwhile, the apoptotic assay revealed that apoptotic rates were dramatically increased in ESCC cells whose NME4 were silenced in comparison with control (Fig. 1f). Transwell assay showed that invasive capabilities of ESCC cells were seriously suppressed relative to control (Fig. 1g). Together, all the data we presented above explicitly indicate that NME4 can promote the proliferation and invasion of ESCC cells.

NME4 promoting proliferation and invasion in ESCC cell lines TE-1 and EC9706. a Baseline expression of endogenous NME4 on mRNA level, as detected by qRT-PCR technique, in three different kinds of ESCC cell lines we enrolled in our setting. GAPDH, internal loading control; one-way ANOVA (Bonferroni) was taken to analyze the difference among the three groups. b Silencing effect, as determined by western-blot, of ESCC cell lines TE-1 and EC9706 transfected with lentiviral-based shRNA-NME4 and its corresponding control, respectively. A total of 50 μg proteins were loaded, the relative variation of NME4 was normalized to GAPDH, acted as internal loading control, c quantitative analysis of variation of NME4 expression shown in western-blot. Two-tailed, unpaired T-test with Welch’s correction was used (T = 10.30, df = 2.112, P value = 0.0077; T = 25.59, df = 2.031, P value = 0.0014), and d proliferative variations, as assayed by MTT method, after NME4 was stably silenced in TE-1 and EC9706 cells. Of note, the error bars on the graphs were too small to be seen here. **P < 0.001 compared with control using the independent sample T-test. e In parallel with MTT assays, proliferative variations, as determined by colony formation assay, after inhibition of NME4. The exact P scores labeled were calculated using two-tailed, unpaired T-test with Welch’s correction. f Likewise, apoptotic variations were analyzed using flow cytometry in TE-1 and EC9706 cells whose NME4 were stably knocked down. The exact P values labeled were calculated using two-tailed, unpaired T-test with Welch’s correction. g Invasive variations were assessed using Transwell assays in TE-1 and EC9706 cells whose NME4 were silenced. The exact P values labeled were calculated using two-tailed, unpaired T-test with Welch’s correction
Differential metabolic products screened out after NME4 were stably knocked down
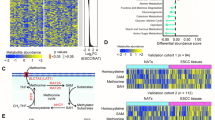
Having understood the oncogenic traits of NME4 in ESCC cells, we next attempted to explore the involvement of NME4 in metabolism of cell lines derived from ESCC that has not been clear yet. To profile the differential metabolites of ESCC cells after TE-1 and EC9706 cells whose endogenous NME4 were stably silenced, widely targeted metabolomic screening strategy was taken (Supplementary Fig. 1). Expectantly, a great deal of differential metabolites, up to 346 kinds of metabolic products (Supplementary Table 1), were obtained. In spite of these numerous differential metabolites that were eventually screened, it turned out that there were only 11 metabolites that were significantly differential between experimental group and control, with 7 metabolites being upregulated and 4 downregulated (Table 1), after cluster analyses performed (Fig. 2a, b). To further comb out these metabolites that were significantly differential, KEGG (Kyoto Encyclopedia of Genes and Genomes) analyses were carried out. KEGG analyses showed that, NME4 were mainly predicative to be involved in metabolic pathways (Fig. 2c). Specifically, the 11 differential metabolites identified comprise Inosine, Hypoxanthine-9-β-d-Arabinofuranoside, 15-oxoETE [15-oxo-5Z,8Z,11Z,13E-eicosatetraenoic acid], 2-Picolinic Acid, B-Nicotinamide Mononucleotide, Nicotinic Acid, 6-Methylnicotinamide, Lumichrome, Pantothenol, Pipecolinic Acid and 12,13-EpOME [(±)12(13)epoxy-9Z-octadecenoic acid] (Fig. 3). These metabolites, if biochemically classified, fundamentally belong to the nucleotide metabolism (Inosine, Hypoxanthine-9-β-d-Arabinofuranoside and B-Nicotinamide Mononucleotide), followed by CoOthersEnzyme Factor and vitamin (Nicotinic Acid and Pantothenol), then came the Oxidized lipid (15-oxoETE and 12, 13-EpOME) and other metabolism in terms of quantity (Table 1).

Cluster and KEGG analyses of the differential metabolites identified after stable silencing of NME4 in ESCC cells. a Cluster analyses, as shown in the form of heat map, of the differential metabolites that were significant after downregulation of NME4 in ESCC cells. In heat map we presented here, on which the products indexed from top to bottom denote MEDP116, 2-Picolinic Acid; MEDN444, Hypoxanthine-9-β-d-Arabinofuranoside; MEDN164, Inosine; MEDP246, Nicotinic Acid; MEDN771, 15-oxoETE [15-oxo-5Z,8Z,11Z,13E-eicosatetraenoic acid]; MEDP249, Pantothenol; MEDP358, Pipecolinic Acid; MEDP890, Lumichrome; MEDN767, 12,13-EpOME [(±)12(13)epoxy-9Z-octadecenoic acid], b bar chart of fold change of metabolic products, indexed totally the same as those in heat map and c Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of differentially metabolic products. Y-axis represents the metabolic signaling pathways that were possibly involved; X-axis denotes the ratio of the number of the metabolic products obtained to the total number of metabolic products annotated in the signaling pathway

Violin plots of metabolic products we screened out that were significantly differential between control group (denoted as N) and experimental group (represented by E). The differential metabolic products indexed MEDP821, 6-Methylnicotinamide; MEDP116, 2-Picolinic Acid; MEDN164, Inosine; MEDN444, Hypoxanthine-9-β-d-Arabinofuranoside; MEDN771, 15-oxoETE [15-oxo-5Z,8Z,11Z,13E-eicosatetraenoic acid]; MEDP246, Nicotinic Acid; MEDP890, Lumichrome; MEDP180, B-Nicotinamide Mononucleotide; MEDN767, 12,13-EpOME [(±)12(13)epoxy-9Z-octadecenoic acid]; MEDP249, Pantothenol; MEDP358, Pipecolinic Acid, respectively
Discussion
In the current investigation, the oncogenic trait of NME4 was firstly defined in vitro in cell lines derived from ESCC in our experimental setting, displaying that NME4 was able to promote the proliferation and invasion of ESCC cells. Metabolically, NME4 was found to be chiefly implicated in the metabolic signaling pathway, outside of nucleotide metabolism. To our knowledge, this is the first report defining the oncogenic traits of NME4 and profiling the differential metabolites after NME4 was stably silenced in ESCC cells.
NME4, also called NM23D, NM23H4, Nm23-H4, or NDPK-D, belongs to the group I members of NME family [7] that can be divided in two groups in mammals according to phylogenetic analyses [8]. Group I comprised NME1, NME2, NME3 and NME4, which share from 58 to 88% identity and are all catalytically active, as systematically reviewed by Lacombe et al. [7]. Unlike the NME1 [9], NME2 [10] or NME3 [11] that have been extensively studied in cancers from different tissues and organs, by contrast, only few data have been available regarding NME4 in human cancers. The original report mentioning Nm23-H4 in cancer came from neuroblastoma [12] where Nm23-H4 was just incidentally mentioned, without much attention being paid. Almost at the same time, in another study concerning diffuse astrocytoma [13], in which Nm23-H4 was also acknowledged in the differential cDNAs that were screened. Later on, in another study about colorectal and renal carcinoma [14], Nm23-H4 was shown to be over-expressed on mRNA level; giving a cue that Nm23-H4 might be oncogenic in colorectal and renal cancer. One subsequent study performed on protein level in human gastric and colon cancer [15], congruently supported that Nm23-H4 protein was over-expressed in neoplasmic tissues compared with normal control, suggesting that Nm23-H4 may be involved in the oncogenesis of gastric and colon cancer. Despite these implicit suggestions from earlier studies, no explicit causal relationship between NME4 or Nm23-H4 and carcinogenesis has been established until the very recent study [3] appeared. In the study [3], lentivirus-mediated knock-down of NME4 was reported to be able to inhibit the abilities of proliferation and colony formation, suggestive of the tumor-promoting role in non-small cell lung cancer; which was in agreement with our observation described here.
Although available evidence acknowledged NME4 as a key player in bioenergetics and lipid signaling and an important contributor to the quality control of the mitochondria (reviewed in [7]), little has been known regarding the differential metabolites mediated by NME4 in cancer cells. Given this, attempt has been made using the widely targeted metabolomic screening to work out the differential metabolites in ESCC cells whose NME4 were stably knocked down compared with control, showing that there were 11 metabolites identified that were significantly differential between silencing of NME4 and control group. Notably, 2-picolinic acid and nicotinic acid whose absolute folds were shown to be far greater than others among the differential metabolites, totaling seven, that were identified to be upregulated. Having identified these differential metabolites, we reasoned that NME4 could negatively regulate these metabolites identified to be upregulated in our setting, which warrants further investigation and confirmation in clinical samples. Biochemically, 2-picolinic acid identified here was a pyridine derivative that involved in kynurenine pathway of tryptophan metabolism [16]. Albeit the role of 2-picolinic acid in ESCC cells was unclear, recent evidence emerged suggesting that picolinic acid has much bearing on immune modulation in cancers [16, 17]. The role picolinic acid mediates in the modulation of tumor immunity within ESCC remains elusive that needs to be further investigated. Nicotinic acid, also called vitamin PP or nicotinamide, much like 2-picolinic acid, biochemically, was implicated in the kynurenine pathway that is the principal route of l-tryptophan catabolism eventually giving birth to the picolinic acid and nicotinamide [18]. Despite the nicotinic acid has been known for decades, its immunomodulatory role in ESCC remains obscure that needs to be further studied.
Among the four metabolites identified to be downregulated with the knock-down of NME4 in ESCC cells, pipecolinic acid has to be mentioned and discussed here because of its most conspicuous downregulation. In pathophysiological environment, little has been described concerning pipecolinic acid. By contrast, the role of pipecolinic acid in immunity has been uncovered gradually with recent works surrounding pipecolinic acid in immunomodulation, which mainly focused on plant, rarely on human. In a more recent study [19], pipecolinic acid was mechanistically found to confer systemic immunity by regulating free radicals in plant. This observation inevitably leads to a speculation that pipecolinic acid may, as in plant, work in the similar way in human cancers. Future investigations on pipecolinic acid in tumor immunity are required to address the speculation.
NME4 mainly located in mitochondria where NME4 have been proposed to work to synthesize ATP or nucleoside diphosphate (NTP) for protein and nucleic acid synthesis [20]. Among its related pathways implicated was nucleotides metabolism, which was expectantly confirmed by our KEGG analyses revealing that Tryptophan, Riboflavin, Purine, Nicotinate, lysine degradation, and Linoleic acid metabolism were also involved in addition to the well-established nucleotides metabolism. The observation we made here was reminiscent of one recent review underscoring that Tryptophan metabolism was associated with cancer immunomodulation [21], therefore can be used as a potential therapeutic target in cancers. Our study presented here back up the current view [22, 23] hold that metabolism, particularly occurred in mitochondria, was associated with tumor immunity. Despite these, there were still two limitations inherent in our study described here. For one thing, all the conclusions drawn on the basis of two kinds of ESCC cell lines in vitro, should have been extended to clinical samples; secondly, differential metabolites identified in our setting warrants further confirmation using the different ESCC cell lines with larger sample size.
In conclusion, NME4 was shown to promote the proliferation and invasion of ESCC cells in vitro. Metabolically, with the aid of widely targeted metabolomic screening, differential metabolites, totaling 11, were identified including Inosine, Hypoxanthine-9-β-d-Arabinofuranoside, 15-oxoETE [15-oxo-5Z,8Z,11Z,13E-eicosatetraenoic acid], 2-Picolinic Acid, B-Nicotinamide Mononucleotide, Nicotinic Acid, 6-Methylnicotinamide, Lumichrome, Pantothenol, Pipecolinic Acid and 12,13-EpOME [(±)12(13)epoxy-9Z-octadecenoic acid]. Some of these metabolites were suggested to be associated with tumor immunomodulation. Our data described above support the relationship between metabolisms occurred in mitochondrial and tumor immunity.
References
Boissan M, Schlattner U, Lacombe ML (2018) The NDPK/NME superfamily: state of the art. Lab Investig 98:164–174. https://doi.org/10.1038/labinvest.2017.137
Boissan M, Dabernat S, Peuchant E, Schlattner U, Lascu I, Lacombe ML (2009) The mammalian Nm23/NDPK family: from metastasis control to cilia movement. Mol Cell Biochem 329:51–62. https://doi.org/10.1007/s11010-009-0120-7
Wang W, Dong M, Cui J, Xu F, Yan C, Ma C, Yi TW, Dong J, Wei Y (2019) NME4 may enhance nonsmall cell lung cancer progression by overcoming cell cycle arrest and promoting cellular proliferation. Mol Med Rep 20:1629–1636. https://doi.org/10.3892/mmr.2019.10413
Liu L, Li M, Zhang C, Zhang J, Li G, Zhang Z, He X, Fan M (2018) Prognostic value and clinicopathologic significance of nm23 in various cancers: a systematic review and meta-analysis. Int J Surg 60:257–265. https://doi.org/10.1016/j.ijsu.2018.10.035
Wang QH, Han W, Chen MB, Bao J, Wang BF (2018) Prognostic value of NM23 in patients with gastric cancer: a systematic review and meta-analysis. J Cancer Res Ther 14:S556–S564. https://doi.org/10.4103/0973-1482.183188
Milon L, Rousseau-Merck MF, Munier A, Erent M, Lascu I, Capeau J, Lacombe ML (1997) nm23-H4, a new member of the family of human nm23/nucleoside diphosphate kinase genes localised on chromosome 16p13. Hum Genet 99:550–557. https://doi.org/10.1007/s004390050405
Lacombe ML, Tokarska-Schlattner M, Boissan M, Schlattner U (2018) The mitochondrial nucleoside diphosphate kinase (NDPK-D/NME4), a moonlighting protein for cell homeostasis. Lab Investig 98:582–588. https://doi.org/10.1038/s41374-017-0004-5
Lacombe ML, Milon L, Munier A, Mehus JG, Lambeth DO (2000) The human Nm23/nucleoside diphosphate kinases. J Bioenerg Biomembr 32:247–258. https://doi.org/10.1023/a:1005584929050
Han W, Shi CT, Cao FY, Cao F, Chen MB, Lu RZ, Wang HB, Yu M, He DW, Wang QH, Wang JF, Xu XX, Ding HZ (2016) Prognostic value of NME1 (NM23-H1) in patients with digestive system neoplasms: a systematic review and meta-analysis. PLoS ONE 11:e0160547. https://doi.org/10.1371/journal.pone.0160547
Thakur RK, Yadav VK, Kumar P, Chowdhury S (2011) Mechanisms of non-metastatic 2 (NME2)-mediated control of metastasis across tumor types. Naunyn Schmiedebergs Arch Pharmacol 384:397–406. https://doi.org/10.1007/s00210-011-0631-0
Yang T, Chen BZ, Li DF, Wang HM, Lin XS, Wei HF, Zeng YM (2016) Reduced NM23 protein level correlates with worse clinicopathologic features in colorectal cancers: a meta-analysis of pooled data. Medicine (Baltim) 95:e2589. https://doi.org/10.1097/MD.0000000000002589
Negroni A, Venturelli D, Tanno B, Amendola R, Ransac S, Cesi V, Calabretta B, Raschella G (2000) Neuroblastoma specific effects of DR-nm23 and its mutant forms on differentiation and apoptosis. Cell Death Differ 7:843–850. https://doi.org/10.1038/sj.cdd.4400720
Huang H, Colella S, Kurrer M, Yonekawa Y, Kleihues P, Ohgaki H (2000) Gene expression profiling of low-grade diffuse astrocytomas by cDNA arrays. Cancer Res 60:6868–6874
Hayer J, Engel M, Seifert M, Seitz G, Welter C (2001) Overexpression of nm23-H4 RNA in colorectal and renal tumours. Anticancer Res 21:2821–2825
Seifert M, Welter C, Mehraein Y, Seitz G (2005) Expression of the nm23 homologues nm23-H4, nm23-H6, and nm23-H7 in human gastric and colon cancer. J Pathol 205:623–632. https://doi.org/10.1002/path.1724
Heng B, Lim CK, Lovejoy DB, Bessede A, Gluch L, Guillemin GJ (2016) Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget 7:6506–6520. https://doi.org/10.18632/oncotarget.6467
Kimura Y, Negishi H, Matsuda A, Endo N, Hangai S, Inoue A, Nishio J, Taniguchi T, Yanai H (2018) Novel chemical compound SINCRO with dual function in STING-type I interferon and tumor cell death pathways. Cancer Sci 109:2687–2696. https://doi.org/10.1111/cas.13726
Adams S, Teo C, McDonald KL, Zinger A, Bustamante S, Lim CK, Sundaram G, Braidy N, Brew BJ, Guillemin GJ (2014) Involvement of the kynurenine pathway in human glioma pathophysiology. PLoS ONE 9:e112945. https://doi.org/10.1371/journal.pone.0112945
Wang C, Liu R, Lim GH, de Lorenzo L, Yu K, Zhang K, Hunt AG, Kachroo A, Kachroo P (2018) Pipecolic acid confers systemic immunity by regulating free radicals. Sci Adv 4:eaar4509. https://doi.org/10.1126/sciadv.aar4509
Gordon DM, Lyver ER, Lesuisse E, Dancis A, Pain D (2006) GTP in the mitochondrial matrix plays a crucial role in organellar iron homoeostasis. Biochem J 400:163–168. https://doi.org/10.1042/BJ20060904
Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA (2019) Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 18:379–401. https://doi.org/10.1038/s41573-019-0016-5
Mills EL, Kelly B, O’Neill LAJ (2017) Mitochondria are the powerhouses of immunity. Nat Immunol 18:488–498. https://doi.org/10.1038/ni.3704
Sukumar M, Kishton RJ, Restifo NP (2017) Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol 46:14–22. https://doi.org/10.1016/j.coi.2017.03.011
Acknowledgements
The study was partly supported by Nature Science Foundation of China (NSFC.81960552, 81960527, 81960527, U1603284, 81860511), and partly by Science and Technology Projects of Xinjiang Uygur Autonomous Region (2018E02067), by Tianshan Xuesong Project of the Xinjiang Uygur Autonomous Region (2018XS19).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there was no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zheng, S., Liu, T., Liu, Q. et al. Widely targeted metabolomic analyses unveil the metabolic variations after stable knock-down of NME4 in esophageal squamous cell carcinoma cells. Mol Cell Biochem 471, 81–89 (2020). https://doi.org/10.1007/s11010-020-03768-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03768-w