
Abstract
Background
Activation of the insulin-like growth factor 1 receptor (IGF-1R)-mediated Janus kinase (JAK)1/2-Stat3 pathway contributes to hepatocarcinogenesis. Specifically, a previous study showed that IGF-1R inhibition downregulated Midkine expression in hepatocellular carcinoma (HCC).
Aims
The present study investigated the role of IGF-1R-JAK1/2-Stat3 and Midkine signaling in HCC, in addition to the molecular link between the IGF-1R-Stat3 pathway and Midkine.
Methods
The expression levels of IGF-1R, Stat3, and Midkine were measured using reverse transcription-quantitative PCR, following which the association of IGF-1R with Stat3 and Midkine expression was evaluated in HCC. The molecular link between the IGF-1R-Stat3 pathway and Midkine was then investigated in vitro before the effect of IGF-1R-Stat3 and Midkine signaling on HCC growth and invasion was studied in vitro and in vivo.
Results
IGF-1R, Stat3, and Midkine mRNA overexpressions were all found in HCC, where the levels of Stat3 and Midkine mRNA correlated positively with those of IGF-1R. In addition, Midkine mRNA level also correlated positively with Stat3 mRNA expression in HCC tissues. IGF-1R promoted Stat3 activation, which in turn led to the upregulation of Midkine expression in Huh7 cells. Similarly, Midkine also promoted Stat3 activation through potentiating JAK1/2 phosphorylation. Persistent activation of this Stat3-Midkine-Stat3 positive feedback signal loop promoted HCC growth and invasion, the inhibition of which resulted in significant antitumor activities both in vitro and in vivo.
Conclusions
Constitutive activation of the IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop is present in HCC, the inhibition of which can serve as a potential therapeutic intervention strategy for HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) comprises ~ 75–85% of all liver cancer cases, which currently ranks sixth in terms of incidence and is the fourth leading cause of cancer-associated mortality worldwide. In 2018, GLOBOCAN reported 841,000 new liver cancer cases and 782,000 deaths as a result of liver cancer annually [1]. In total, ~ 50% all HCC cases occur in China, where the 5-year survival rate is only ~ 12% due to the lack of accurate early diagnostic methods [2, 3]. Patients who are diagnosed with HCC at an advanced stage stand little chance of benefitting efficaciously from surgical resection. In addition, other treatment options, including local ablation, transarterial chemoembolization or intra-arterial infusion chemotherapy, and transplantation in various combinations, confer prognostic benefits only for a limited number of patients with unresectable advanced HCC [4,5,6,7]. Therefore, development of novel molecular-targeted therapies against HCC may provide additional avenues to improve the success of treatment [8]. Sorafenib, a multikinase inhibitor that blocks Raf, vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF) receptor signaling, is the most commonly used molecular-targeted drug to treat HCC, but it only minimally enhances patient survival. Therefore, better treatment options are still needed [9].
Insulin-like growth factor 1 receptor (IGF-1R) is a transmembrane tyrosine kinase that is expressed in a wide variety of cell types. The IGF-1R gene is located on the 15q25–26 locus, which is ~ 100 kb in length and contains 21 exons. IGF-1R is a heterodimer that is comprised of two identical α-subunits and equal β-subunits, where the former induces the autophosphorylation of tyrosine residues on the latter upon activation [10]. The phosphorylated intracellular β-subunits then trigger an intracellular signaling cascade that has been documented to regulate cell survival and proliferation [11, 12]. Under physiological conditions, IGF-1R mediates the biological functions of insulin-like growth factor 1(IGF-1) and IGF-2 to modulate metabolism, embryonic development, and cellular differentiation [13, 14]. Recent studies have shown that IGF-1R overexpression is strongly associated with the malignancy of various cancers such as HCC [15,16,17,18,19], suggesting IGF-1R inhibition to be a promising therapeutic target for HCC [20].
It has been demonstrated previously that IGF-1R-mediated signaling pathways in human malignancies mainly involve PI3K/Akt, MAPK, and the Janus kinase (JAK) 1/2-Stat3 [21,22,23,24]. However, the precise downstream targets of the IGF-1R-JAK1/2-Stat3 pathway remain to be fully elucidated (Fig. 1). A previous study revealed that IGF-1R inhibition by RNA interference (RNAi) can significantly suppress HCC growth and invasion by downregulating Midkine expression, suggesting that IGF-1R positively regulates Midkine in HCC [25]. Therefore, in the present study, the functional role of the IGF-1R-JAK1/2-Stat3 pathway and Midkine signaling in HCC was investigated further, with focus on the molecular link between the IGF-1R-Stat3 pathway and Midkine activity.

IGF-1R-mediated signaling pathways. IGF-1 (or IGF-2) binding to IGF-1R promotes intrinsic tyrosine kinase activity and autophosphorylation, which can recruit and phosphorylate the adaptor proteins IRS1/2 and Shc. The interaction of IRS1/2 with IGF-1R induces the activation of PI3K/AKT and consequently the disinhibition of mTOR oncoprotein. In parallel, Shc activation induces the activation of the RAS/RAF/MAPK pathway. Another signaling cascade downstream of IGF‑1R is the JAK1/2-Stat3 pathway, where direct activation of Janus kinase1/2 occurs, leading to the activation and phosphorylation of Stat3 molecule; Stat3 further activates the expression of the Midkine gene that may result in the growth and invasion in hepatocellular carcinoma cells
Materials and Methods
Cell Lines
The human liver cancer cell lines (Huh7, Hep3B, HCCLM3), a human immortalized liver cell line (THLE-2), and a 293 T cell line were purchased from American Type Culture Collection and cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc., catalog number 11960044) supplemented with heat-inactivated 10% FBS (Gibco; Thermo Fisher Scientific, Inc., catalog number 10270). All cell lines were maintained in a 37˚C incubator under 5% CO2.
Human Tissue Specimens
Tumor tissue specimens and matched adjacent nontumor tissues (MANT) of 32 patients with HBV infection-positive HCC (sex, 25 male and seven female) were obtained from the Biological Sample Bank of the First Affiliated Hospital of Jinan University (Guangzhou, China). The diagnosis of HCC was made by pathological examination, and none of the patients received any preoperative treatment prior to admission. An additional control group of 13 patients (sex, 11 male and two female) with liver rupture (ten cases) and hepatic hemangioma (three cases) were recruited, of whom normal adult liver tissue (NALT) samples were also obtained from the Biological Sample Bank of the First Affiliated Hospital of Jinan University. HBV infection was defined as positive detection of serum HBsAg. The present study was approved by the medical ethics committee of the First Affiliated Hospital of Jinan University, and written informed consent was obtained from each participant.
Reverse Transcription-Quantitative PCR (RT-qPCR)
Total RNA was extracted from the cell lines, human hepatic tissue specimens, and tumor tissues isolated from mice using the TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc., catalog number 15596016) according to manufacturer’s protocol. RNA was then reverse-transcribed to obtain cDNA using ten units of Reverse Transcriptase XL (AMV) (TaKaRaBio, Inc., catalog number D2620) according to the manufacturer’s protocols. qPCR was then performed using the Power SYBR® Green PCR Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc., catalog number 4309155). The sequences of the primers used for RT-qPCR are listed in Table S1.
Western Blot Analysis
Total protein was extracted from the human HCC cell lines using RIPA buffer (Pierce; Thermo Fisher Scientific, Inc., catalog number 89900). Equal amounts of protein for each sample were subjected to SDS-PAGE followed by transferring to nitrocellulose membranes (Bio-Rad Laboratories, Inc., catalog number 16201). The membranes were first incubated with antibodies against IGF-1R (Abcam, catalog number ab39675), Midkine (Abcam, catalog number ab52637), phosphorylated Stat3 (Cell Signaling Technology, Inc., catalog number 9145), Stat3 (Cell signaling technology, Inc., catalog number 49999904 T), JAK1 (Abcam, catalog number ab47435), phosphorylated JAK1 (Abcam, catalog number ab47337), JAK2 (Abcam, catalog number ab39636), phosphorylated JAK2 (Abcam, catalog number ab32101), and GAPDH (Abcam, catalog number ab9484), before incubation with horseradish peroxidase-conjugated secondary antibodies. GAPDH was served as the loading control.
Enzyme-Linked Immunosorbent Assay
The concentration of Midkine was measured in the cell culture supernatants by an enzyme-linked immunosorbent assay (ELISA) using a commercial kit (Abcam, catalog number ab193761). The assay was performed according to the manufacturer’s instructions.
Construction of the Vectors Expressing IGF-1R, Stat3, and Midkine
The coding region cDNAs of human IGF-1R (NM_000875.5), Stat3 (NM_139276.2), and Midkine (NM_001270551.2) were cloned by reverse transcription from mRNA isolated from Huh7 cells. They were then cloned into the pcDNA3.1( +) vector (Invitrogen; Thermo Fisher Scientific, Inc., catalog number V79020) according to the manufacturer’s protocol, to obtain pcDNA3.1( +)-IGF-1R, pcDNA3.1( +)-Stat3, and pcDNA3.1( +)-Midkine vectors. The primers used for the construction of the aforementioned vectors are listed in Table S1.
RNA Interference
Huh7 cells with stable knockdown of IGF-1R expression (shIGF-1R) and their corresponding negative control (shCon) were constructed by transfecting Huh7 cells with the lentivirus-encoded IGF-1R short hairpin RNA (shRNA) and lentivirus-negative control shRNA in procedures that were described previously [25]. For the transient knockdown of Stat3 and Midkine expression, Huh7 cells were transfected with small interfering RNAs (siRNAs) targeting human Stat3 and Midkine using Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc., catalog number 13778–075), respectively. The sequences of siRNAs/shRNAs used in the present study for IGF-1R knockdown (shIGF-1R), negative control (shCon), Stat3 siRNA (siStat3), and Midkine siRNA (siMidkine) are listed in Table S1.
Construction of Luciferase Reporter Plasmids
The promoter region of the Midkine gene (GenBank accession no. 4192) was analyzed using the JASPAR database (http://jaspar.genereg.net), where nine putative Stat3 binding sites were found. Among them, three Stat3 binding sites (Sites 1–3) with the highest scores were selected for further validation (Table S2). The Midkine promoter was cloned by PCR from the DNA isolated from Huh7 cells into the HindIII and NcoI restriction sites of the pGL3 promoter vector (Promega Corporation, catalog number E1761). The recombinant luciferase reporter construct encoding the wild-type (WT) Midkine promoter was termed pGL3-Midkine-WT. The luciferase reporter constructs encoding each of the mutated (MUT) sequences of the three Stat3 binding sites aforementioned on the Midkine promoter were generated based on that of the pGL3-Midkine-WT using a site-directed mutagenesis kit (Stratagene; Agilent Technologies, Inc., catalog number 200519). They were termed as pGL3-Midkine-MUT. The primers used for the generation of recombinant luciferase reporter constructs are listed in Table S1. All constructs were verified by sequencing.
Cell Transfection and Dual-Luciferase Reporter Assay
Huh7 cells were plated at a density of 2 × 105 cells/well into a 24-well dish and transiently cotransfected with each of the luciferase reporter constructs and the pRL-TK vector (Promega Corporation, catalog number E2241) for the normalization of transfection efficiency, using TransFast™ transfection reagent, according to manufacturer’s protocol (Promega Corporation, catalog number E2431). Luciferase assays were performed according to the manufacturer’s protocol 48 h after transfection.
Quantitative Chromatin Immunoprecipitation (qChIP) Assay
Huh7 cells were transfected with either the pcDNA3.1( +)-Stat3 vector or siRNA oligonucleotides specific for Stat3. qChIP assay was then performed using a ChIP assay kit according to the manufacturer’s protocols (Upstate Biotechnology, Inc., catalog number 17–295) 72 h after transfection. Briefly, the cells were cross-linked with 1% formaldehyde for 10 min at 37˚C before being stopped by incubation with glycine at room temperature for 5 min. The cells collected were then washed twice with ice-cold PBS containing a protease inhibitor cocktail (Roche Applied Science, catalog number 04693159001) resuspended in SDS lysis buffer (Upstate Biotechnology, Inc., catalog number 20–163) before being subjected to sonication. Each sample was subsequently precleared by incubation with protein G agarose and divided into three subsamples and allocated as follows: i) Subsample 1 was used as an input control; ii) subsample 2 was incubated with anti-Stat3 (Cell signaling technology, Inc., catalog number 49999904 T) antibodies; and iii) subsample 3 was incubated with the nonspecific rabbit IgG antibody (negative control, Santa Cruz Biotechnology, Inc., catalog number sc-2004) overnight at 4˚C. DNA purified from both the immunoprecipitated and input samples was then subjected to qPCR analysis using Power SYBR® Green PCR Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc., catalog number 4309155). The primers used for ChIP assays are listed in Table S1.
Cell Viability Assay
To measure cell viability, cells were seeded into 96-well culture plates at a density of 1 × 104 cells per well. At 0, 24, 48, and 72 h after culture, 20 μl 5 mg/ml MTT (Sigma-Aldrich, Merck KGaA, catalog number M2128) was added to each well and the plates were incubated at 37˚C for 4 h. The formazan crystals were then lysed in DMSO and the optical density values were measured at 490 nm in each well.
Transwell Cell Migration/Invasion Assays
Uncoated Transwell chambers (pore size, 8 μm; Corning, Inc., catalog number 3428) and those coated with Matrigel (BD Biosciences, catalog number 356234) were used in migration and invasion assays, respectively. Matrigel was added into the upper chambers and incubated at 37˚C incubator for 2 h before use. The cells were first seeded into the upper chambers, following which Transwell assays were performed according to the manufacturer’s protocols. After the Transwell chambers were incubated for 24 h at 37˚C in a humidified atmosphere under 5% CO2, the lower chamber was stained with crystal violet. Cell migration/invasion was evaluated by counting the cells that had migrated/invaded through the pore.
Preparation of Lentiviral Constructs and Lentivirus Production
Lentiviral constructs encoding IGF-1R, Stat3, and Midkine were prepared by using the pLVX-IRES-ZsGreen1vectors (Clontech Laboratories, Inc., catalog number 632187) according to the manufacturer’s protocols. Briefly, the coding regions of human IGF-1R, Stat3, and Midkine genes were cloned into the pLVX-IRES-ZsGreen1vector to obtain the pLVX-IGF-1R, pLVX-Stat3, and pLVX-Midkine constructs. In addition, sequences of shRNA targeting the human Stat3 gene were synthesized and cloned into this lentiviral vector to produce the pLVX-shRNA2 vector (Clontech Laboratories, Inc., catalog number 632179), which was named as pLVX-shStat3. These constructs were then packaged into lentiviral particles using 293 T cells by applying the Lenti-X™ HTX Packaging System (Clontech Laboratories, Inc., catalog number 631249) to produce lentiviral particles termed lenti-IGF-1R, lenti-Stat3, lenti-Midkine, and lenti-shStat3, respectively. At 48 h after transfection, the lentiviral particles were harvested and stored at -80˚C for further use. The sequence of the shStat3 used for the present study is listed in Table S1.
HCC Xenograft Tumor Models in Nude Mice
BALB/c nude mice (weight, 27–34 g; age, 6 weeks) were purchased from the Guangdong Medical Laboratory Animal Center (Guangzhou, China). A total of 48 male BALB/c nude mice were randomly divided into eight groups (groups A-H; n = 6 mice per group). The mice were subcutaneously injected with 4 × 106 Huh7 cells transfected with the lentivirus-negative control shRNA [Huh7-shRNA(-)] into the right flank in groups A-C or Huh7 cells transfected with the lentivirus encoding IGF-1R shRNA [Huh7-shRNA( +)] in groups D-H.
On day10 after injection, tumors were observed on the nude mice. The mice in group A were injected with normal saline into the tumor, whereas those in group B were injected with ~ 6 × 107 copies of lenti-IGF-1R. Mice in group C were injected with ~ 6 × 107 copies of lenti-Midkine in combination with lenti-shStat3, whilst those in group D were injected with normal saline. Those in group E were injected with ~ 6 × 107 copies of lenti-Stat3, and mice in group F were injected with ~ 6 × 107 copies of lenti-Midkine. Group G received injections with ~ 6 × 107 copies of lenti-shStat3, whereas mice in group H were injected with ~ 6 × 107 copies of lenti-Midkine in combination with lenti-shStat3. Following injection, tumor growth was monitored. The largest (A) and smallest (B) superficial diameters of the tumors were measured using Vernier calipers, where the tumor volume (mm3) was calculated using the following formula: A × B2 × 0.5. All animal experiments were approved by the Medical Ethics committee of the First Affiliated Hospital of Jinan University and performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the First Affiliated Hospital of Jinan University.
Statistical Analysis
Categorical data were compared using Chi-squared or Fisher’s exact tests, while quantitative data were analyzed using the independent-samples t test or one-way ANOVA followed by Mann–Whitney U test or Kruskal–Wallis test. Correlation was evaluated by Pearson’s correlation coefficient analysis. SPSS 21.0 software (IBM Corp.) was used for all statistical analyses. All results were obtained from three independent experiments performed in triplicate. P < 0.05 was considered to indicate a statistically significant difference.
Results
mRNA Expression of IGF-1R, Stat3, and Midkine Is Upregulated in HCC Cells and Human HCC Tissues
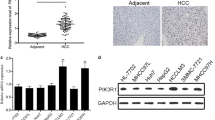
To measure the expression of IGF-1R, Stat3, and Midkine, we performed RT-qPCR, where it was found that the expression of IGF-1R mRNA, Stat3 mRNA, and Midkine mRNA was significantly upregulated in the HCC cell lines Huh7, Hep3B, HCCLM3 compared with that in the normal liver cell line THLE-2 (Fig. 2a).

The overexpression of IGF-1R, Stat3, and Midkine is found in HCC cells and human HCC tissues. a The mRNA expression of IGF-1R, Stat3, and Midkine was detected by quantitative RT-PCR in HCC cells (Huh7, Hep3B, and HCCLM3) and a normal liver cell (THLE-2). The results were shown as means ± SD and compared by independent-samples t test or one-way ANOVA. b The mRNA expression of IGF-1R, Stat3, and Midkine was detected by quantitative RT-PCR in human HCC tissues (HCC) (n = 32) and their matched adjacent nontumorous tissues (MANT) (n = 32), and normal adult liver tissues (NALT) (n = 13). The results were compared by Kruskal–Wallis H test. c Linear correlation analyses between IGF-1R mRNA and the mRNA expression of Stat3 and Midkine, and between Stat3 mRNA and Midkine mRNA in HCC tissues (n = 32). *P < 0.05; **P < 0.01
Subsequently, the human liver tissue specimens were examined and RT-qPCR analysis revealed that expression of IGF-1R mRNA, Stat3 mRNA, and Midkine mRNA was also significantly upregulated in human HCC tissues compared with that in MANT and NALT, respectively (Fig. 2b). In particular, IGF-1R mRNA expression was also found to be higher in MANT compared with that in NALT (Fig. 2b). Further correlation analysis demonstrated that the expression levels of Stat3 mRNA (r = 0.716, P < 0.01) and Midkine mRNA (r = 0.681, P < 0.01) were positively correlated with those of IGF-1R mRNA in HCC tissues (Fig. 2c). Additionally, the levels of Midkine mRNA expression were also found to correlate positively with those of Stat3 mRNA in HCC tissues (r = 0.657, P < 0.01; Fig. 2c).
Furthermore, in 32 HCC samples, the levels of IGF-1R mRNA, Stat3 mRNA, and Midkine mRNA for the cases with tumor embolus of portal vein (TEPV) and lymph node metastasis, and with poorly differentiated HCC tissues (Edmondson grade III–IV) were significantly higher than those for cases without TEPV and lymph node metastasis, and with well-differentiated HCC tissues (Edmondson grade I-II), respectively. However, no correlation was found between the levels of IGF-1R mRNA, Stat3 mRNA, and Midkine mRNA and the remaining clinicopathological parameters including age, a-fetoprotein level, tumor size, and tumor nodularity (P > 0.05) (Table 1). These results seem to suggest the possibility that the expression levels of IGF-1R mRNA, Stat3 mRNA, and Midkine mRNA may be positively associated with the more malignant behavior of HCC. In addition, the clinical characteristics of 32 HCC patients and 13 patients with hepatic rupture (HR) and hepatic hemangioma (HH) are presented in Table S3.
IGF-1R promotes Midkine expression by upregulating Stat3 expression and inducing Stat3 protein phosphorylation in HCC cells. To further explore the effect of IGF-1R on the expression of Stat3 and Midkine, an IGF-1R overexpression vector was transiently transfected into Huh7 cells. RT-qPCR results suggested that the overexpression of IGF-1R increased the mRNA expression of Stat3 and Midkine (Fig. 3a). Increased expression of the Stat3 and Midkine proteins and Stat3 phosphorylation by IGF-1R overexpression was subsequently confirmed by western blot analysis in Huh7 cells compared with those in the blank control (Fig. 3b). By contrast, stable knockdown of IGF-1R by RNAi reduced the mRNA expression of Stat3 and Midkine (Fig. 3a), in addition to reducing the expression of Stat3 protein, Stat3 phosphorylation, and Midkine protein in Huh7 cells compared with those in the blank control (Fig. 3b). These observations suggest that IGF-1R positively regulates the expression of both Stat3 and Midkine in HCC cells.

Effect of IGF-1R on the expression of Stat3 and Midkine, and effect of Stat3 on Midkine expression in HCC cells. a, b The expression of Stat3 and Midkine at both mRNA (a) and protein (b) levels was detected by quantitative RT-PCR and western blot, respectively, in Huh7 cells with stable IGF-1R knockdown (shIGF-1R), Huh7 cells with transient overexpression of IGF-1R (IGF-1R), Huh7 cells with both stable IGF-1R knockdown and transient overexpression of Stat3 (shIGF-1R + Stat3), and the blank control (Bla) (untreated Huh7 cells). c The concentration of Midkine was measured in the cell culture supernatants by ELISA in Huh7 cells with stable IGF-1R knockdown (shIGF-1R), Huh7 cells with transient overexpression of IGF-1R (IGF-1R), Huh7 cells with both stable IGF-1R knockdown and transient overexpression of Stat3 (shIGF-1R + Stat3), and the blank control (Bla) (untreated Huh7 cells). d, e The expression of Midkine at both mRNA (d) and protein (e) levels was detected by quantitative RT-PCR and western blot, respectively, in Huh7 cells with transient overexpression of Stat3 (Stat3), Huh7 cells with transient Stat3 knockdown (siStat3), and the blank control (Bla) (untreated Huh7 cells). f Dual-luciferase assays in Huh7 cells cotransfected with the firefly luciferase constructs containing the WT or MUT Midkine promoter and Stat3 expression vector (Stat3) or the negative control vector (NC). g The binding of Stat3 to the top 3 Stat3 binding sites of the Midkine promoter was detected by qChIP assay in Huh7 cells with transient Stat3 knockdown (siStat3), Huh7 cells with transient overexpression of Stat3 (Stat3), or the blank control (Bla) (untreated Huh7). The results were shown as means ± SD and compared by independent-samples t test or one-way ANOVA. **P < 0.01; N.S., no significant difference
To investigate the effects of Stat3 on Midkine expression, Stat3 overexpression vector was transiently transfected into Huh7 cells with IGF-1R expression stably knocked down by RNAi. Transient overexpression of Stat3 markedly reversed the suppression of Midkine expression on both mRNA and protein levels induced by IGF-1R silencing (Fig. 3a and b). Additionally, We also evaluated the secreted Midkine by ELISA assay in Huh7 cells with genetic manipulation of shIGF-1R, IGF-1R overexpression, and shIGF-1R + STAT3. The results showed that ectopic overexpression of IGF-1R (IGF-1R) increased Midkine secretion, whereas stable knockdown of IGF-1R (shIGF-1R) decreased Midkine secretion in Huh7 cells compared with the blank control (Bla). Moreover, transient overexpression of Stat3 in Huh7 cells with stable knockdown of IGF-1R expression (shIGF-1R + Stat3) markedly rescued inhibition of Midkine secretion induced by silencing IGF-1R (Fig. 3c). On the other hand, compared with that in the blank control, transient overexpression of Stat3 was found to increase the expression of Midkine mRNA and protein in Huh7 cells, which was reduced by RNAi-mediated transient Stat3 knockdown (Fig. 3d and e). Taken together, these results suggest that IGF-1R upregulates Midkine expression by increasing Stat3 activity in HCC cells.
Upregulation of Midkine Expression by Stat3 Involves the Binding of Stat3 to the Midkine Promoter in HCC Cells
To further explore the mechanism by which Stat3 upregulates Midkine expression, a total of nine potential Stat3 binding sites in the Midkine promoter were predicted using the JASPAR database, following which the top three scoring potential Stat3 binding sites were selected for further validation using luciferase assay. The Midkine promoter containing the wild-type or mutant DNA sequences of the three aforementioned Stat3 binding sites was then cloned into the pGL3 basic vectors, to produce the pGL3-Midkine-WT and pGL3-Midkine-MUT reporters, respectively. These reporters were subsequently cotransfected into Huh7 cells with either the Stat3 overexpression vector or the negative control vector. Compared with cells transfected with the negative control, cotransfection with the Stat3 plasmid induced a ~ 1.7-fold increase in the relative luciferase activity of the pGL3-Midkine-WT reporter but not in the pGL3-Midkine-MUT reporter, where the relative luciferase activity remains unaffected (Fig. 3f).
ChIP assay was next performed to verify whether Stat3 binds to the Midkine promoter in Huh7 cells. Transfection with siStat3 by RNAi led to the reduced enrichment of Stat3 on the three potential Stat3 binding sites of the Midkine promoter but the transient overexpression of Stat3 resulted in the increased enrichment of Stat3 on these three Stat3 binding sites compared with that in the blank control (Fig. 3g). These observations indicate that Stat3 upregulates Midkine expression by binding to the Midkine promoter in HCC cells.
Midkine Promotes Stat3 Activation in a Positive Feedback Manner in HCC Cells
In the present study, the expression of Midkine mRNA was positively correlated with Stat3 expression in HCC tissues, suggesting that Midkine may also regulate Stat3 expression. According to this notion, the effect of Midkine on Stat3 expression was next explored. Midkine was transiently overexpressed in Huh7 cells with IGF-1R expression stably knocked down. Transient overexpression of Midkine was found to significantly reverse the inhibition of Stat3 mRNA and protein expression (but not IGF-1R), in addition to reversing the inhibition of Stat3 phosphorylation induced by stable IGF-1R knockdown compared with those in the blank control (Fig. 4a and b). By contrast, transient knockdown of Midkine expression by RNAi reduced the expression of Stat3 mRNA, Stat3 protein, and Stat3 phosphorylation, in addition to lowering JAK1/2 phosphorylation expression, while transient Midkine overexpression increased the expression of Stat3 mRNA, Stat3 protein, and Stat3 phosphorylation, along with potentiating JAK1/2 phosphorylation expression in Huh7 cells compared with those in the blank control (Fig. 4c and d); moreover, transient knockdown or overexpression of Midkine did not affect the expression of JAK1/2 mRNA, JAK1/2 protein, and IGF-1R mRNA and protein (Fig. 4c and d) in Huh7 cells (Fig. 4c and d). These results suggest that Midkine is also able to promote Stat3 activation by JAK1/2-Stat3 signaling in a positive feedback manner in HCC cells. Collectively, the data presented in this study suggest that there is an constitutive activation of the IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop in HCC.

Effect of Midkine on Stat3 expression in HCC cells. a, b The expression of Stat3 at both mRNA (a) and protein (b) levels was detected by quantitative RT-PCR and western blot, respectively, in Huh7 cells with stable IGF-1R knockdown (shIGF-1R), Huh7 cells with both stable IGF-1R knockdown and transient overexpression of Midkine (shIGF-1R + Midkine), and the blank control (Bla) (untreated Huh7). c, d The expression of IGF-1R, JAK1, JAK2, and Stat3 at both mRNA (c) and protein (d) levels was detected by quantitative RT-PCR and western blot, respectively, in Huh7 cells with transient Midkine knockdown (siMidkine), Huh7 cells with transient overexpression of Midkine (Midkine), and the blank control (Bla) (untreated Huh7). The results were shown as means ± SD and compared by independent-samples t test or one-way ANOVA. **P < 0.01; N.S., no significant difference
IGF-1R Promotes HCC Cell Proliferation, Migration, and Invasion by Activating the Stat3-Midkine-Stat3 Positive Feedback Signaling Loop
To evaluate the effect of persistent IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop activation on the biological behavior of HCC cells, MTT assay and Transwell assays were performed. Transient overexpression of IGF-1R in Huh7 cells significantly enhanced HCC cell viability (Fig. 5), migration (Fig. 6a and b), and invasion (Fig. 6c and d), while stably knocking down IGF-1R expression in Huh7 cells by shIGF-1R transfection was found to reduce HCC cell viability (Fig. 5) and inhibit migration (Fig. 6a and b) and invasion (Fig. 6c and d) compared with those in the blank control. In addition, the combination of transient Stat3 knockdown with the transient overexpression of Midkine in Huh7 cells did not affect HCC cell viability (Fig. 5), migration (Fig. 6a and b) and invasion (Fig. 6c and d) compared with those in the blank control (Bla). By contrast, transient overexpression of Stat3 or Midkine in IGF-1R-deficient Huh7 cells markedly reversed the inhibitory effects of stable IGF-1R knockdown on HCC cell viability (Fig. 5), migration (Fig. 6a and b), and invasion (Fig. 6c and d). Transient knockdown of Stat3 expression in Huh7 cells significantly potentiated the inhibition mediated by stable IGF-1R knockdown on HCC cell viability (Fig. 5), migration (Fig. 6a and b) and invasion (Fig. 6c and d) compared with those in cells with stable IGF-1R knockdown alone. Additionally, combined Stat3 knockdown and Midkine overexpression in Huh7 cells deficient in IGF-1R expression did not affect HCC cell viability (Fig. 5), migration (Fig. 6a and b), or invasion (Fig. 6c and d) compared with cells IGF-1R with stable IGF-1R knockdown alone. These findings suggest that IGF-1R increases HCC cell viability, migration, and invasion by triggering the Stat3-Midkine-Stat3 positive feedback loop, whereas inhibition of this loop significantly suppressed the growth and invasive capabilities of HCC cells.

Effect of the persistent activation of IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop on HCC cell proliferation. Cell proliferation was evaluated by MTT assay, and the results were expressed by absorbance at 490 nm. and compared by independent-samples t test or one-way ANOVA. **P < 0.01; N.S., no significant difference

Effect of the persistent activation of IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop on HCC cell migration and invasion. a, b Cell migration was evaluated by transwell assay, and the results were expressed by the number of migrated cells and compared by independent-samples t test or one-way ANOVA. c, d Cell invasion was evaluated by transwell assay, and the results were expressed by the number of invaded cells and compared by independent-samples t test or one-way ANOVA. *P < 0.05; **P < 0.01; N.S., no significant difference
IGF-1R Promotes HCC Cell-Derived Xenograft Tumor Growth in Nude Mouse Through Triggering the Stat3-Midkine-Stat3 Positive Feedback Signal Loop
To investigate the in vivo relevance of the aforementioned in vitro findings, the effects of persistent IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop activation on HCC cell-derived xenograft tumorigenicity were next investigated. Nude mice were subcutaneously injected with 4 × 106Huh7-shRNA(-) cells in groups A-C or Huh7-shRNA( +) cells in groups D-H. Tumors were then formed 10 days after inoculation before experiments were performed by injections into the tumor. Compared with mice in group A, which were treated with normal saline, tumor growth was found to be significantly higher in mice in group B, which were treated with lenti-IGF-1R and significantly lower in mice in group D, which were injected with Huh7-shRNA( +) cells but were treated with normal saline (Fig. 7a–c). Tumor growth was not markedly affected in mice in group C, which were treated with the combination of lenti-shStat3 and lenti-Midkine, compared with mice in group A (Fig. 7a–c). Correspondingly, compared with that in mice in group A, the mRNA expression of IGF-1R, Stat3, and Midkine was found to be significantly increased in the tumor tissues of mice in group B, while that in mice in group D was found to be significantly reduced (Fig. 8a–c). No significant difference was found in the mRNA expression of IGF-1R, Stat3, and Midkine in the tumor tissues of mice in group C compared with that in mice in group A (Fig. 8a–c).

Effect of the persistent activation of IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop on tumorigenesis in vivo. a Tumors from the tumor-bearing nude mice 35 days after inoculation. b, c Tumor sizes were measured and tumor volumes were calculated. The results were shown as means ± SD and compared by independent samples t test or one-way ANOVA. *P < 0.05; N.S., no significant difference

Effect of the persistent activation of IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop on the mRNA expression of IGF-1R, Stat3, and Midkine of xenograft tumor tissues in nude mouse. a–c The expression of IGF-1R (a), Stat3 (b), and Midkine (c) was detected by quantitative RT-PCR in xenograft tumor tissues from the nude mice. The results were shown as means ± SD and compared by independent-samples t test or one-way ANOVA. **P < 0.01; N.S., no significant difference
Compared with mice in group D, which were injected with Huh7-shRNA( +) cells, inhibition of tumor growth by stable IGF-1R knockdown was revealed to be markedly reversed in mice in group E, which were treated with lenti-Stat3 and in mice in group F, which were treated with lenti-Midkine (Fig. 7a–c). In addition, compared with that in mice in group D, tumor growth was inhibited further in mice in group G, which were treated with lenti-shStat3, while tumor growth was not markedly affected in mice in group H, which were treated with lenti-shStat3 and lenti-Midkine combined (Fig. 7a–c). Consistent with these observations, compared with that in tumor tissues from mice in group D, mRNA expression of Stat3 and Midkine in the tumor tissues of mice in groups E or F was found to be significantly increased, whereas that in mice in group G was significantly reduced (Fig. 8b and c). There were no significant differences in the mRNA expression levels of Stat3 and Midkine in mice in group H nor in IGF-1R mRNA expression in mice in groups E–H compared with those in tumor tissues from mice in group D (Fig. 8a–c). These findings suggest that IGF-IR promotes HCC cell-derived xenograft tumor growth in nude mouse by activating the Stat3-Midkine-Stat3 positive feedback loop, whereas inhibition of this loop exhibited significant antitumor activities in vivo.
Discussion
HCC is a common and aggressive malignancy that is associated with dreadful outcomes worldwide [9]. The carcinogenesis of HCC is highly complex, where the molecular mechanism of pathogenesis remains poorly understood [27]. Therefore, it is necessary to uncover the underlying mechanism to establish a superior therapeutic approach for HCC. IGF-1R is a transmembrane receptor tyrosine kinase that can activate the MAPK, PI3K/Akt, and JAK1/2-Stat3 signaling pathways (Fig. 1) and is activated by IGF-1 and IGF-2 [28, 29]. Many studies have reported that IGF-1R overexpression is strongly associated with a number of cancers such as HCC [30,31,32,33] and the activation of the IGF-1R-Stat3 signaling pathway has been reported to contribute to the development of cancers including HCC [24, 26, 28]. However, downstream targets of this pathway in HCC remain to be fully elucidated.
Stat3 belongs to the Stat family of transcription factors that has been reported to regulate a wide range of cellular processes, including cell proliferation, differentiation, apoptosis, angiogenesis, immune responses, tumorigenesis, and cancer metastasis [26, 28, 34, 35]. In addition, it has been previously demonstrated that Stat3 is essential for the establishment of cell transformation and cancer development by regulating the transcription of a number of key genes [26, 28]. Midkine is a heparin-binding growth factor that was originally reported to be the product of a retinoic acid-responsive gene during embryogenesis [36]. The expression levels of Midkine are high during embryogenesis but become negligible and restricted in healthy human adult tissues [37, 38]. Recent studies have suggested that Midkine is implicated in the development of human cancers, including breast, bladder, colon cancer, and HCC [39,40,41,42].
The present study investigated the role of IGF-1R-Stat3 and Midkine signaling in the development of HCC and the molecular link between the IGF-1R-Stat3 signaling pathway and Midkine. It was demonstrated that the mRNA expression of IGF-1R, Stat3, and Midkine was markedly increased in the HCC cells and the human HCC tissues. Correlation analysis showed that the mRNA levels of Stat3 and Midkine were positively correlated with those of IGF-1R mRNA in the HCC tissues. In addition, Midkine mRNA levels were also found to be positively correlated with those of Stat3 mRNA in the HCC tissues. Moreover, It was found that the HCC patients with the higher mRNA levels of IGF-1R, Stat3, and Midkine were more often associated with TEPV, lymph node metastasis, and poor tumor differentiation (Edmondson grade III–IV). Consistent with results from the present study, several studies have previously demonstrated increased IGF-1R expression in HCC [25, 32, 33]. He et al.[43] reported that the activity of Stat3 was increased in HCC tissues [43], while Calvisi et al.[44] also previously found that the expression of Stat3 in HCC tissues was significantly higher compared with that in normal liver and paracancerous tissues [44]. Dai et al.[45] demonstrated that the expression of Midkine mRNA and protein was markedly increased in human HCC tissues [45]. Taken together, these aforementioned findings suggest that the IGF-1R-Stat3 pathway and Midkine expression are activated in HCC, which may be associated with the more malignant behavior of HCC.
The association between the IGF-1R-Stat3 signaling pathway and Midkine molecule was subsequently investigated. The present study found that IGF-1R overexpression promoted Stat3 expression and Stat3 activation, which in turn led to the upregulation of Midkine expression in Huh7 cells. Further mechanistic investigation demonstrated that Stat3 increased Midkine expression by binding to the promoter of the Midkine gene in Huh7 cells. By contrast, Midkine was also able to conversely promote Stat3 activation through potentiating JAK1/2 phosphorylation. Taken together, these findings support the notion that there is a persistently activated Stat3-Midkine-Stat3 positive feedback signal loop mediated by IGF-1R activation in HCC, where IGF-1R contributes to the development of HCC by activating this signaling loop.
It has been previously demonstrated that the IGF-1R-JAK1/2-Stat3 signaling cascade is implicated in embryonic development, cell transformation, and tumor development [28]. Under physiologic condition, this pathway is activated by growth factors, which results in the activation of Stat target genes, including protein inhibitors of activated Stats, SH2-containing phosphatases, and suppressors of cytokine signaling (SOCS) [44, 46,47,48]. These inhibitory proteins, with SOCS1 and SOCS3 serving as previously reported examples, can inhibit IGF-1R-JAK1/2-Stat3 signaling by binding to activated JAK1/2 to inhibit their catalytic activity. In this manner, this signaling cascade can be tightly regulated by the negative feedback loop mediated by these specific inhibitors [28, 49]. However, under pathological conditions such as carcinogenesis, this pathway becomes persistently activated due to the suppression of these inhibitors. Inactivation of SOCS1 has been frequently found in malignant tumors such as HCC to constitutively activate the JAK1/2-Stat3 pathway [44, 50,51,52]. However, the molecular mechanism underlying the suppression of JAK1/2-Stat3 inhibitors in HCC development remains unclear, which requires further investigation.
To investigate the role of the IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback loop in the development of HCC further, in vitro and in vivo experiments were performed. Activation of this signal loop was found to increase Huh7 cell viability, migration, and invasion and potentiate Huh7 cell-derived xenograft tumorigenesis in nude mice. Conversely, inhibition of this signaling loop significantly suppressed the growth and invasive capabilities of Huh7 cells and Huh7 cell-derived xenograft tumor growth. Notably, it was also found that the simultaneous inhibition of both IGF-1R and Stat3 expression in this signaling cascade significantly suppressed HCC growth and invasion in an additive manner, implicating the synergistic antitumor activity of this method, which can serve as a potential therapeutic strategy for HCC.
In conclusion, to the best of our knowledge, the present study demonstrated for the first time the existence of the constitutively active IGF-1R-mediated Stat3-Midkine-Stat3 positive feedback signal loop in human HCC. This is different from a previous report, which found that the uncontrolled activation of JAK1/Stat3 signaling was due to the suppression of specific inhibitory proteins in human HCC [44]. The molecular mechanism responsible for the persistent activation of this signaling pathway in liver cancer requires further investigation. Increased IGF-1R expression contributes to HCC growth and invasion at least partially through the activation of this positive feedback loop, the inhibition of which demonstrated significant antitumor activities, suggesting this to be a potential approach for the gene therapy of HCC.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- IGF-1:
-
Insulin-like growth factor 1
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- JAK1/2:
-
Janus kinase ½
- MAPK:
-
Mitogen-activated protein kinase
- MANT:
-
Matched adjacent nontumorous tissues
- MTT:
-
Methyl thiazolyl tetrazolium
- NALT:
-
Normal adult liver tissues
- PI3K:
-
Phosphatidyl-inositol-3 kinase
- qChIP:
-
Quantitative chromatin immunoprecipitation
- PIAS:
-
Protein inhibitors of activated Stats
- RNAi:
-
RNA interference
- SHP:
-
SH2-containing phosphatases
- shRNA:
-
Short hairpin RNA
- siRNAs:
-
Small interfering RNAs
- SOCS:
-
Suppressors of cytokine signaling
- Stat3:
-
Signal transducer and activator of transcription 3
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424
Chen W, Zheng R, Baade PD et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132
Lee SS, Shin HS, Kim HJ et al. Analysis of prognostic factors and 5-year survival rate in patients with hepatocellular carcinoma: a single-center experience. Korean J Hepatol. 2012;18:48–55
Morise Z, Kawabe N, Tomishige H et al. Recent advances in the surgical treatment of hepatocellular carcinoma. World J Gastroenterol. 2014;20:14381–14392
Lim C, Shinkawa H, Hasegawa K et al. Salvage liver transplantation or repeat hepatectomy for recurrent hepatocellular carcinoma: an intent-to-treat analysis. Liver Transplant. 2017;23:1553–1563
Kondo Y, Kimura O, Shimosegawa T. Radiation therapy has been shown to be adaptable for various stages of hepatocellular carcinoma. World J Gastroenterol. 2015;21:94–101
Huang SX, Wu YL, Tang CW et al. Prophylactic hepatic artery infusion chemotherapy improved survival after curative resection in patients with hepatocellular carcinoma. Hepatogastroenterology. 2015;62:122–125
Ladju RB, Pascut D, Massi MN et al. Aptamer: A potential oligonucleotide nanomedicine in the diagnosis and treatment of hepatocellular carcinoma. Oncotarget. 2017;9:2951–2961
Ogunwobi OO, Harricharran T, Huaman J, Galuza A, Odumuwagun Q, Tan Y, Ma GX, Nguyen MT. Mechanisms of hepatocellular carcinoma progression. World J Gastroenterol. 2019;25:2279–2293
Girnita L, Worrall C, Takahashi SI et al. Something old, something new and something borrowed: emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell Mol Life Sci. 2014;71:2403–2427
Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34
LeRoith D, Werner H, Beitner-Johnson D, Roberts CT. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163
Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–518
Werner H. For debate: the pathophysiological significance of IGF-I receptor overexpression: new insights. Pediatr Endocrinol Rev. 2009;7:2–5
Singh P, Alex JM, Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol. 2014;31:805
Adachi Y, Yamamoto H, Ohashi H et al. A candidate targeting molecule of insulin-like growth factor-I receptor for gastrointestinal cancers. World J Gastroenterol. 2010;16:5779–5789
Yeo CD, Park KH, Park CK et al. Expression of insulin-like growth factor 1 receptor (IGF-1R) predicts poor responses to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer patients harboring activating EGFR mutations. Lung Cancer. 2015;87:311–317
Sun WY, Yun HY, Song YJ et al. Insulin-like growth factor 1 receptor expression in breast cancer tissue and mammographic density. Mol Clin Oncol. 2015;3:572–580
Nakajima N, Kozu K, Kobayashi S et al. The expression of IGF-1R in Helicobacter pylori-infected intestinal metaplasia and gastric cancer. J Clin Biochem Nutr. 2016;59:53–57
Ryan PD, Goss PE. The emerging role of the insulin-like growth factor pathway as a therapeutic target in cancer. Oncologist. 2008;13:16–24
Kamrava M, Gius D, Casagrande G, Kohn E. Will targeting insulin growth factor help us or hurt us?: An oncologist’s perspective. Ageing Res Rev. 2011;10:62–70
Delafontaine P, Song YH, Li Y. Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arterioscler Thromb Vasc Biol. 2004;24:435–444
Kim WY, Prudkin L, Feng L et al. Epidermal growth factor receptor and K-Ras mutations and resistance of lung cancer to insulin-like growth factor 1 receptor tyrosine kinase inhibitors. Cancer. 2012;118:3993–4003
Subramani R, Lopez-Valdez R, Arumugam A et al. Targeting Insulin-Like Growth Factor 1 Receptor Inhibits Pancreatic Cancer Growth and Metastasis. PLoS One. 2014;9:e97016
Bie CQ, Liu XY, Cao MR et al. Lentivirus-mediated RNAi knockdown of insulin-like growth factor-1 receptor inhibits the growth and invasion of hepatocellular carcinoma via down-regulating Midkine expression. Oncotarget. 2016;7:79305–79318
Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers. 2014;6:926–957
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576
Zong CS, Chan J, Levy DE, Horvath C, Sadowski HB, Wang LH. Mechanism of STAT3 activation by insulin-like growth factor I receptor. J Biol Chem. 2000;275:15099–15105
Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol. 2007;19:117–123
Belinsky MG, Rink L, Cai KQ et al. The insulin-like growth factor system as a potential therapeutic target in gastrointestinal stromal tumors. Cell Cycle. 2008;7:2949–2955
Steller MA, Delgado CH, Bartels CJ, Woodworth CD, Zou Z. Overexpression of the insulin-like growth factor-1 receptor and autocrine stimulation in human cervical cancer cells. Cancer Res. 1996;56:1761–1765
Kim SO, Park JG, Lee YI. Increased expression of the insulin-like growth factor I (IGF-I) receptor gene in hepatocellular carcinoma cell lines: implications of IGF-I receptor gene activation by hepatitis B virus X gene product. Cancer Res. 1996;56:3831–3836
Tovar V, Alsinet C, Villanueva A et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J Hepatol. 2010;52:550–559
Sanchez-Lopez E, Flashner-Abramson E, Shalapour S et al. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 Receptor-insulin receptor substrate and STAT3 signalingscript. Oncogene. 2016;35:2634–2644
Wu J, Du J, Fu X et al. Iciartin, a novel FASN inhibitor, exerts anti-melanoma activities through IGF-1R/STAT3 signaling. Oncotarget. 2016;7:51251–51269
Muramatsu T. Midkine: a promising molecule for drug developmentto treat diseases of the central nervous system. Curr Pharm Des. 2011;17:410–423
Luo J, Wang X, Xia Z et al. Transcriptional factor specificity protein 1 (SP1) promotes the proliferation of glioma cells by up-regulating midkine (MDK). Mol Biol Cell. 2015;26:430–439
Kadomatsu K, Muramatsu T. Midkine and pleiotrophin in neural development and cancer. Cancer Lett. 2004;204:127–143
Ibusuki M, Fujimori H, Yamamoto Y et al. Midkine in plasma as a novel breast cancer marker. Cancer Sci. 2009;100:1735–1739
Muramaki M, Miyake H, Hara I, Kamidono S. Introduction of midkine gene into human bladder cancer cells enhances their malignant phenotype but increases their sensitivity to antiangiogenic therapy. Clin Cancer Res. 2003;9:5152–5160
Krzystek-Korpacka M, Diakowska D, Grabowski K, Gamian A. Tumor location determines midkine level and its association with the disease progression in colorectal cancer patients: a pilot study. Int J Colorectal Dis. 2012;27:1319–1324
Koide N, Hada H, Shinji T et al. Expression of the midkine gene in human hepatocellular carcinomas. Hepatogastroenterology. 1999;46:3189–3196
He G, Yu GY, Temkin V et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297
Calvisi DF, Ladu S, Gorden A et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117–1128
Dai LC, Yao X, Lu YL et al. Expression of midkine and its relationship with HBV infection in hepatocellular carcinomas. Zhonghua yi xue za zhi. 2003;83:1691–1693
Wormald S, Hilton DJ. Inhibitors of cytokine signal transduction. J Biol Chem. 2004;79:821–824
Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19:378–387
Kile BT, Alexander WS. The suppressors of cytokine signalling (SOCS). Cell Mol Life Sci. 2001;58:1627–1635
Starr R, Hilton DJ. Negative regulation of the JAK/STAT pathway. Bioessays. 1999;21:47–52
Fukushima N, Sato N, Sahin F, Su GH, Hruban RH, Goggins M. Aberrant methylation of suppressor of cytokine signalling-1 (SOCS-1) gene in pancreatic ductal neoplasms. Br J Cancer. 2001;89:338–343
Nagai H, Kim YS, Lee KT et al. Inactivation of SSI-1, a JAK/STAT inhibitor, in human hepatocellular carcinomas, as revealed by two-dimensional electrophoresis. J Hepatol. 2001;34:416–421
Yoshikawa H, Matsubara K, Qian GS et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet. 2001;28:29–35
Acknowledgments
The authors appreciate the patients, study investigators, and staff who participated in this study.
Funding
This study was supported by Grants from Shenzhen Science and Technology Project of China (No. JCYJ20160428100513152); Science and Technology Funding Project of Hunan Province, China (No. 2017SK4010); 7th batch Key Laboratory Project of Colleges and Universities of Hunan Province (2019–301); Medical Science and Technology Research Fund of Guangdong Province (No. A2019256); Natural Science Foundation of Guangdong Province (No. 2018A0303130302); and Medical Science and Technology Foundation of Guangdong Province (No. A2018011).
Author information
Authors and Affiliations
Contributions
S.H.T. designed the study. C.Q.B., Y.F.C., H.J.T., Q.L., L.Z., X.J.P., S.Y., J.Q.L., J.L.L., and S.L.W. performed the experiments. C.Q.B., Y.F.C., H.J.T., and Q.L. conducted statistical analysis. C.Q.B., Y.F.C., and S.H.T. wrote the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Ethics approval and consent to participate
The study was approved by the medical ethics committee of the First Affiliated Hospital of Jinan University, China. Written consent was obtained from all participants. This study conforms to the Declaration of Helsinki.
Consent for publication
Written informed consent for publication was obtained from the patients. All authors have agreed to publish this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bie, C., Chen, Y., Tang, H. et al. Insulin-Like Growth Factor 1 Receptor Drives Hepatocellular Carcinoma Growth and Invasion by Activating Stat3-Midkine-Stat3 Loop. Dig Dis Sci 67, 569–584 (2022). https://doi.org/10.1007/s10620-021-06862-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-021-06862-1