
Abstract
Insulin and insulin-like growth factor (IGF) signaling system, commonly known for fine-tuning numerous biological processes, has lately made its mark as a much sought-after therapeutic targets for diabetes and cancer. These receptors make an attractive anticancer target owing to their overexpression in variety of cancer especially in prostate and breast cancer. Inhibitors of IGF signaling were subjected to clinical cancer trials with the main objective to confirm the effectiveness of these receptors as a therapeutic target. However, the results that these trials produced proved to be disappointing as the role played by the cross talk between IGF and insulin receptor (IR) signaling pathways at the receptor level or at downstream signaling level became more lucid. Therapeutic strategy for IGF-1R and IR inhibition mainly encompasses three main approaches namely receptor blockade with monoclonal antibodies, tyrosine kinase inhibition (ATP antagonist and non-ATP antagonist), and ligand neutralization via monoclonal antibodies targeted to ligand or recombinant IGF-binding proteins. Other drug-discovery approaches are employed to target IGF-1R, and IR includes antisense oligonucleotides and recombinant IGF-binding proteins. However, therapies with monoclonal antibodies and tyrosine kinase inhibition targeting the IGF-1R are not evidenced to be satisfactory as expected. Factors that are duly held responsible for the unsuccessfulness of these therapies include (a) the existence of the IR isoform A overexpressed on a variety of cancers, enhancing the mitogenic signals to the nucleus leading to the endorsement of cell growth, (b) IGF-1R and IR that form hybrid receptors sensitive to the stimulation of all three IGF axis ligands, and (c) IGF-1R and IR that also have the potential to form hybrid receptors with other tyrosine kinase to potentiate the cellular transformation, tumorigenesis, and tumor vascularization. This mini review is a concerted effort to explore and fathom the well-recognized roles of the IRA signaling system in human cancer phenotype and the main strategies that have been so far evaluated to target the IR and IGF-1R.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the past few decades, cancer has emerged as a global problem claiming numerous lives, making it a leading cause of mortality second only to cardiovascular disease. Global Cancer Statistics 2011 have reported breast cancer and lung cancer to be the most frequently diagnosed cancers in females and males, respectively, as well as the leading cause of cancer death. Prostate cancer is the second most frequently diagnosed visceral cancer and the sixth leading cause of cancer death among males [1]. The hypothetical premise that cancer and diabetes are associated, having been investigated extensively in both in vitro and in vivo experimental models, had been confirmed by numerous studies highlighting the association of diabetes to an increased risk of several types of cancer including the prostate and breast cancers [2–5]. Recently published data have led the researchers to reconsider and reinterpret the previous results as diabetes is a complex metabolic disorder characterized by hyperglycemia, hyperinsulinemia, obesity, and lipidemia [6, 7]. Recent epidemiological studies have also confirmed that type 2 diabetes mellitus (T2DM) is associated with an increased risk for many forms of cancer, including cancer of the breast, colon, liver, pancreas, and kidney [8–12]. In vivo and in vitro experimental models have highlighted increased insulin, IGF-1 and IGF-2 signaling to be responsible for enhancing tumorigenesis [13–15]. Insulin, IGF-1 and IGF-2 signaling through the cognate or hybrid receptor can induce tumorigenesis, accounting to some extent for the link between diabetes mellitus (DM) and cancer [16–18]. The metabolic syndrome including obesity, hypertension, dyslipidemia, and hyperglycemia is related to insulin resistance and hyperinsulinemia [19–21]. These factors are hallmarks of DM and cancer, resulting in increased DM-associated cancer risk. A variety of approaches to target insulin receptor (IR) and IGF-1R, inclusive of antisense oligonucleotides, antisense expression plasmids, IGF-binding proteins, ATP antagonist small-molecule kinase inhibitors, non-ATP antagonist small-molecule kinase inhibitors, and neutralizing antibodies, have been utilized to study IR and IGF-1R signaling [22–26]. Among the strategies mentioned above, the three most investigated strategies include tyrosine kinase inhibitors, monoclonal antibodies, and neutralizing antibodies. Tyrosine kinase inhibitors are orally available and more easily penetrate the blood–brain barrier owing to their small-molecular size, whereas monoclonal antibodies are more likely to be specific to the target, avoiding toxicities caused by inhibition of IR and IGF-1R. Wide spectrum covered by the tyrosine kinase inhibitors might have alternative benefits due to the overexpression of IRA and IGF-1R receptor on variety of cancers including the breast, prostate cancer, and human osteoblastogenesis [27–30]. Currently, more than 25 molecules are at different stages of development and used for the clinical and preclinical development of both approaches in cancer.
Physiological roles of insulin and insulin-like growth factor
Insulin and IGF are peptides pivotal to numerous functions such as cellular growth, proliferation, metabolism, glucose homeostasis, cell differentiation, and apoptosis [31]. Insulin is secreted by β-cells of pancreas in response to the increasing blood glucose levels. As a consequence of this response, insulin binds to tyrosine kinase receptors on to the surface of the classic insulin-responsive cells, mainly the hepatocytes, adipocytes, and muscle cells, which express high levels of IR [32]. In addition, IR is also expressed in the tissues including the prostate gland and breast tissue. Insulin, although is primarily involved in regulating metabolism, also influences the normal growth of the cells. Metabolic potential of insulin is responsible for maintaining the metabolic activity and glucose homeostasis but the mitogenic potential increases the tissue-specific cell proliferation in addition to modulating survival and metastasis of the disease [33], thus contributing to increased risk of tissue-specific cancers. On the contrary, signaling of IGF as growth hormone plays a fundamental role in regulating embryonic growth as well as specific differentiation in most adult tissues [34].
Insulin and IGF ligands
Insulin and IGF are peptides having 40–80 % homology making it challenging, although not impossible, to explain insulin and IGF-1 ligand receptor interaction. Insulin/IGF signaling system mainly comprises of three ligands—IGF-1, IGF-2, and insulin, which in turn interact with at least six receptors as represented in the Fig. 1: the type I IGF receptor (IGF-1R), the IRA (IR-A), the IRB (IR-B), hybrid receptors of IGF and IR-A, hybrid receptors of IGF and IR-B, and hybrid receptors of IR-A and IR-B. Insulin when in blood circulation, called insulin ligand, is a monomer consisting of two chains, an α-chain of 21 amino acids and a β-chain of 30 amino acids linked by two disulfide bridges [35]. IGFs are small, single-chain polypeptide ligands (7–8 kD) with an intact C-domain derived from prepropeptides in a manner similar to insulin [36]. The mature IGF-1 and IGF-2 peptides consist of β- and α-domains that are homologous to β- and α-chains of insulin [37]. Furthermore, in the cellular microenvironment, six IGF-binding proteins (IGFBP1–6) are present, which are not only crucial in regulating the bioavailability of IGFs by competing with IGFR and IGFBP proteases but also modulate the balance between IGFs and IGFBPs [38, 39]. IGFBPs and IGFs comprise a major superfamily of protein hormones that regulate mitogenesis, differentiation, survival, and other IGF-stimulated events in both normal and cancerous cells [39, 40]. An in vivo study indicated that IGFBP3 inhibits the tumor growth of HER2 overexpressing human breast cancer cells [41, 42]. Furthermore, it was reported that high expression IGFBP2 was not associated with reduced cell proliferation in breast cancer, glioblastoma, prostate, and ovarian cancer suggesting that IGFBP can affect cell function in an independent manner, although their role in cancer is not yet clear [43–45].

IGF axis is comprised of three ligands—IGF-1, IGF-2, and insulin itself, which interacts with at least six receptors: the type I IGF receptor (IGF-1R), the IRA (IR-A), the IRB (IR-B), hybrid receptors of IGF and IR-A, hybrid receptors of IGF and IR-B, hybrid receptors of IR-A and IR-B. Structurally, all IR and the IGFR have two extracellular α-subunits and two transmembrane β-subunits that are joined to each other by disulfide bonds. Insulin binds with high affinity to IR-A, IR-B, and IGF-1R, and IGF-1 binds to the IGF-1R and to the hybrid receptor IGF-1R/IR-A or IGF-1R/IR-B. IGF-2 binds to IR-A, IGF-1R or to IGF-1R/IR-A hybrid receptor. Insulin and insulin growth factor ligand bind to IGF-1R, IR-A, and hybrid receptors of IGF and IR-A, mediate the mitogenic signaling pathway, while ligands binding to IR-B activate metabolic signaling. Binding to the hybrid receptors, leading to mitogenic or metabolic signaling, is determined by the IR isoform that formed the hybrid receptors
Insulin and IGF receptors and signaling
IR and IGF-1R are heterotetrameric protein consisting of two extracellular α-subunits and two transmembrane β-subunits each [46]. The binding of ligand to α-subunits of IR subsequently stimulates the intrinsic tyrosine kinase activity of the β-subunits of the receptor. These receptor possess the ability to autophosphorylate and transphosphorylate intracellular substrates, as a consequence initiating a cascade of complex cellular responses of intermediate protein. The activated IR tyrosine kinase activates several substrates including IR substrate proteins (IRS1-4), Gab-1, Cbl, and Shc, Phosphatidyl Inositol 3-Kinase (PIK3), Akt, mTOR, MAPK, and signal regulatory protein family [47].
Insulin and IGF hybrid receptors
IGF-1R and IR are overexpressed on variety of cancer including prostate, breast, osteosarcoma, and thyroid carcinomas [48–51], thus leading to the hypothesis that they may be forming hybrid receptors in a variety of cancers. IGF-1R is highly homologous to the IR, sharing 84 % amino acid identity in the kinase domain and 100 % conservation on the ATP binding pocket [52, 53], as a consequence of which IR and IGF-1R proreceptors may heterodimerize to form insulin–IGF hybrid receptors, comprising one α-subunit and one β-subunit each of the IR and IGF-1R. Structurally, all IR and the IGFR have two extracellular α-subunits and two transmembrane β-subunits that are joined by disulfide bonds [18, 33]. Insulin binds with high affinity to IR-A, IR-B, and IGF-1R, IGF-1 have high affinity to the IGF-1R and two hybrid receptor IGF-1R/IR-A or IGF-1R/IR-B. IGF-2 binds to IR-A, IGF-1R or to hybrid receptor IGF-1R/IR-A [54, 55]. These hybrid heterodimeric receptors are known to play a consequential role in receptor signaling in normal and abnormal tissues. Research carried out on human cancer highlighted the fact that autophosphorylation of IR/IGF-1R hybrid receptors in response to insulin and IGF-1 resulted in increased cell proliferation, indicating that hybrid receptors were the major mediators of IGF signaling in these cells [56–59].
Tissue-specific expression of IR
A major insulin target tissue, called insulin-responsive tissue, encompasses liver, adipose tissue, and skeletal muscle. Nevertheless, IR expression is not only constrained to insulin target tissues, but their occurrence has also been established in numerous insulin-unresponsive tissue like the brain, heart, kidney, pulmonary alveoli, pancreatic acini, placenta vascular endothelium, monocytes, granulocytes, erythrocytes, and fibroblasts [60]. This suggests that IR may be functionally playing an inextricable role in the non-metabolic effects in addition to metabolic effects. Metabolic and non-metabolic effects of IR are supported by the effects of insulin on growth and development [61]. In addition, lifestyle, nutrition, and exercise have also been reported to influence IR expression [62]. Co-expression of IR and IGF-1R has been observed to enhance the risk of cancer initiation and progression. IR has two isoform, IR-A and IR-B, where IRA is mitogenic, whereas the later one is metabolic, and the involvement of IRA in primary human prostate and breast cancer has been reported. The role played by IGF-1R and IR in a number of cancers makes them an irrefutable target for cancer treatment. Numerous factors have been held responsible for the aberrant IRA expression known to contribute to the deregulated response of cancer cells brought about by the insulin and IGF, (a) IR overexpression may occur leading to increase in the sensitivity of insulin, thereby increasing the pleiotropic effects of circulating insulin, especially during hyperinsulinemic and insulin resistance. (b) IRA overexpression may bind to IGF-1R and IR-B and form hybrid receptor. These studies taken together with the finding that IR-A is often aberrantly expressed in cancer cells have strengthened the hypothesis that insulin resistance and compensatory hyperinsulinemia are a pivotal link between diabetes and cancer [63].
Strategies to target the IR and IGF-1R in cancer therapy
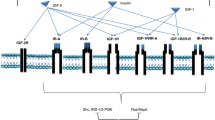
Inhibition of IR and IGF-1R can be achieved as a consequence of sundry experimental approaches that include employing the dominant negative mutants, kinase defective mutants, antisense oligonucleotides, antisense expression plasmids, IGF-binding proteins, soluble forms of the receptor, neutralizing antibodies, and small-molecule tyrosine kinase inhibitors (ATP antagonist and non-ATP antagonist) of the IR and IGF-1R activity [25]. This review mainly seeks to discuss the three approaches currently being evaluated in clinical trials: (A) tyrosine kinase inhibitors, (B) neutralizing antibodies, and (C) IGF-binding proteins. Approaches for therapeutic intervention of IGF-1R and IR are outlined in Fig. 2. Neutralizing antibodies are the monoclonal antibodies developed to target the ectodomain-binding domain of the IR and IGF-1R, which block ligands binding while tyrosine kinase inhibitors compete with ATP in the kinase domain. Tyrosine kinase inhibitors and antibodies that target IGF-1R and IR are tabulated in Table 1. A schematic structure of tyrosine kinase inhibitors is shown in Fig. 3.

Approaches for the therapeutic intervention of IGF-1R and IR. The IGF-1R and IR are hetrotetrameric transmembrane receptor tyrosine kinase formed by two α-and two β-subunits linked by disulfide bonds. The binding of ligand to α-subunits of IGF-1R and IR stimulates the intrinsic tyrosine kinase activity of the β-subunits of the receptor. These receptors have the ability to autophosphorylate and transphosphorylate intracellular substrates proteins that lead to complex cellular responses of cascade of intermediate protein, which contribute to sustain proliferation, inhibit apoptosis, and elicit transformation. The three main approaches are used for IGF-1R and IR inhibition, receptor blockade with monoclonal antibodies, tyrosine kinase inhibition (ATP antagonist and non-ATP antagonist), and ligand neutralization via monoclonal antibodies targeted to ligand or recombinant IGF-binding proteins (IGFBPs)

Examples of ATP antagonist and non-ATP antagonist IGF-IR and IR tyrosine kinase inhibitors
IR and IGF-1R tyrosine kinase inhibitor
BMS-536924
BMS-536924 is the small molecule of ATP-competitive dual IR and IGF-IR kinase inhibitor synthesized by Bristol Myers Squibb that has contributed significantly in inhibiting constitutive receptor phosphorylation as well as downstream signaling through MEK, Akt, and mTOR [64]. As evident in several reports, BMS-535924 is an effective inhibitor of the IR and IGF-1R in vitro, showing an IC50 of 73 and 100 nM, respectively. BMS-535924 administration is also known to inhibit cell proliferation in in vivo experimental tumor model and human tumor xenografts [65–67] as well as it possesses the ability to inhibit cell proliferation and induce apoptosis in a wide variety of human cancer cell lines viz, colon, breast, lung, pancreas, prostate, sarcoma, and multiple myeloma [68]. Interestingly, BMS-536924 does not alter the sensitivity of insulin-stimulated glucose uptake on muscle [69]. HER1 and HER2 receptors are overexpressed in MCF-7 breast cancer cell, showing cross talk with IGF-1R and associated with drug resistance and poor prognosis. Thus, combination or multitargeted studies to target both receptors may prove to be beneficial for advanced metastatic cancers. These results suggest that the combinations of IGF-1R inhibitors with other targeted therapies in clinical studies are important to achieve better patient outcomes [70, 71]. Furthermore, it is also reported that treatment with the combination of BMS-754807 with docetaxel has resulted in improved tumor regression in addition to reduced proliferation and increased apoptosis [72].
BMS-754807
BMS-754807 is an oral small-molecule ATP-competitive inhibitor of the IGF-1R and IR receptor tyrosine kinase, which is a potent inhibitor of the growth of a broad range of cancer cell lines including mesenchymal epithelial and hematopoietic tumor cell lines [73]. The combination of cetuximab and BMS-754807 in vivo resulted in improved clinical outcome as compared to single-agent treatment in dose-dependent manner [74]. Remarkably, BMS-754807 was active in Rh41-MAB391R cells and able to overcome resistance to MAB391, signifying broader clinical activity of BMS-754807 against IR and IGF-1R [75]. BMS-754807 also exhibited antiproliferative effects in vitro in combination with 4-hydroxy tamoxifen and fulvestrant. Moreover, combined treatment of BMS-754807 with either tamoxifen or letrozole in vivo elicited significant tumor regression not achieved by single-agent therapy [76]. IR pathway in IGF-IR null mouse embryonic fibroblasts was hypersensitive to insulin ligand stimulation, and IRA expression level was not changed [77]. BMS-754807 was tested against the Pediatric Preclinical Testing Program (PPTP) cell lines in vitro, and was observed to be reliable with a specific IGF-IR effect that is seen in a minority of the PPTP cell lines. Most consistent in vivo activity was observed among the neuroblastoma, and rhabdomyosarcoma panels [78]. Clinical result also shows that daily dosing of BMS-754807 resulting in exposures exceeding preclinical MEE is feasible and safe [79].
Tyrphostin (AG-1024)
AG-1024 is a specific IGF-1R and IR inhibitor with IC50 of 0.4 and 0.1 μM, respectively. A series of small-molecular compounds known as tyrphostins were reported as tyrosine kinase inhibitor [80, 81]. Tyrphostin AG-1024 is a specific inhibitor of IGF-1R and IR tyrosine kinase activity, which inhibits insulin-stimulated cellular proliferation. Furthermore, the previously reported tyrphostins agent AG490 provides an evidence regarding the application of AG490 in prevention and reversal of autoimmune T1D in NOD mouse [82].
Linsitinib (OSI-906)
OSI-906 is an orally bioavailable small-molecule ATP-competitive, imidazopyrazine-based dual inhibitor of IGF-1R, and IR with IC50 biochemical potency of 35 and 75 nM, respectively. In vitro cell assays reveal that OSI-906 potently inhibits IGF-1R and IR. In vivo efficacy and LISN xenograft model showed a dose-dependent effect on tumor growth inhibition [83]. OSI-906 is a potent, active, and selective small-molecule inhibitor of the IGF-1R signaling axis [84]. OSI-906 was observed to inhibit ligand-dependent autophosphorylation and transphosphorylation of both human IGF-IR and IR in cells, which ultimately resulted in the inhibition of variety of growth-promoting molecules including PI3 K, Akt, and MEK, followed by the inhibition of cell proliferation and stimulation of apoptosis in a variety of tumor cell lines [85]. Interestingly, exceptional selectivity profile of OSI-906 in conjunction with its ability to inhibit both IGF-1R and IR provides the exclusive opportunity to fully target the IGF-1R and IR [86]. Antitumor activity of OSI-906 has been demonstrated in breast cancer [87], ovarian carcinoma [88] lung cancer [89], and colorectal cancer models [90].
BMS-554417
BMS-554417, a novel small molecule developed as an inhibitor of IR/IGF-1R, possessing potential clinical applications owing to their antiproliferative and pro-apoptotic activity in vitro and in vivo. BMS-554417 plays a crucial role in inhibiting IGF-1R and IR kinase activity and cell proliferation in vitro, and reduces tumor xenograft size in vivo, with IC50 120 nmol/L [91].
Picropodophyllotoxin (PPP)
PPP is an orally active IGF-1R inhibitor found to be effective to some extent toward IR, fibroblast growth factor receptors (FGFR), and platelet-derived growth factor receptor (PDGFR). PPP has been currently tested as an orally administrated single agent and in combination with other drugs in advanced cancer patients with solid tumors [92]. PPP has the ability to inhibit IGF-1R autophosphorylation, as a consequence resulting in inhibiting cell survival and up-regulating apoptosis. Furthermore, PPP was also seen to impede growth in multiple myeloma cell lines, bone marrow stromal cells [93], uveal melanoma cells [94], 5T33MM mouse model [95], and colon cancer cells [96].
NVP-AEW541
NVP-AEW541 is a pyrrolo [2,3-d] pyrimidine-derived tyrosine kinase inhibitor with a high selectivity for IGF-1R, with an IC50 of 0.086 μM than IR having an IC50 of 2.3 μM in biological assays. NVP-AEW541 abrogates IGF1-mediated transformation of normal cell to malignant cellular phenotype by the inhibition of IGF-1R signaling. In vivo, this orally bioavailable compound inhibits IGF-1R signaling in tumor xenografts and significantly reduces the growth of IGF-1R-driven fibrosarcomas [97]. Antitumor activity of NVP-AEW541 has been successfully established in musculoskeletal tumors, hepatocellular carcinoma, NET, malignant mesothelioma, HNSCC, colorectal cancer, neuroblastoma, Ewing’s sarcoma, multiple myeloma, pancreatic cancer, gastrointestinal stromal tumor, gastrointestinal cancer, breast cancer, synovial sarcoma, biliary tract cancer, and epithelial ovarian cancer [98–110]. As expected for a specific IGF-1R kinase inhibitor, NVP-AEW541 abrogated IGF1-mediated cell survival and inhibited the cell proliferation of cultured tumor cell lines, inducing apoptosis and cell cycle arrest in vitro. Moreover, NVPAEW541 significantly inhibited the growth of tumor xenografts in vivo [111]. Accumulating support suggests that NVP-AEW541 represents a potential therapeutic strategy for the treatment for variety of tumor types in which IGF-1R is overexpressed.
Furthermore, a related inhibitor, NVP-ADW642, was tested in combination with imatinib, an inhibitor of c-kit in Ewing/s tumor cells. A synergistic effect was seen with dual treatment with NVP-ADW742 and imatinib, which was associated with significant changes in phosphorylation PI3 K and mTOR downstream signaling molecules of IR and IGF-1R [112]. Moreover, in both osteosarcoma and Ewing sarcoma cell lines, trastuzumab has been found to be effective when combined with IGF-1R inhibition [113].
Multitargeted inhibitor
The Di-diabody targeting EGFR and IGF-1R is a novel tetravalent bispecific antibody that is an immunoglobulin-based molecules binding to two different epitopes on either the same or distinct antigens [114–116]. Dual inhibitor Nordihydro guaiaretic Acid (INSM-18) is an orally available small-molecule tyrosine kinase inhibitor that has demonstrated selective inhibition of IGF1 and human EGFR [17]. It has been well established that this inhibitor exhibits potent antitumor activity against breast, lung, pancreatic, and prostate tumors [117]. Two single-dose phase I clinical studies in healthy volunteers have concluded that INSM-18 is safe and a well-tolerated anticancer plant product [118, 119]. XL228 is a protein kinase inhibitor targeting IGF-1R, the AURORA kinases, FGFRI-3, ABL and SRC family kinases. Aurora kinases control crucial steps in mitotic progression and cytokinesis [93]. XL-228 has also been reported to inhibit xenograft models including the MCF7, Colo205, HT29, and A549 in addition to other cancer cell line.
KW-2450
KW-2450 is an orally active, multi-kinase inhibitor, which inhibits both insulin IGF-1R and IR with an IC50 of 7.39 and 5.64 nmol/L, respectively, and also exhibited inhibitory activities against several protein tyrosine kinases such as FAK, FLT1, FLT3, JAK2, KDR, TRKA, and Aurora A in addition to inhibiting the growth of various types of malignant tumors.
IGF-1R monoclonal antibodies
Human monoclonal antibodies target the extracellular domain of IGF-1R and IR resulting in blocking of the IGF-1R/IR-mediated downstream signaling pathways. Numerous antibodies have been developed and successfully established in preclinical and clinical phases.
EM164 (murine AVE1642)
Murine AVE1642 is an antagonistic monoclonal antibody developed by Sanofi-Aventis/Immuno Gen, which was observed to specifically bind to IGF-1R but not to the homologous IR. EM164 bound strongly to IGF-1R with a dissociation constant of 0.1 nM, thereby potently antagonizing the cell proliferation and survival functions of the receptor in cancer cells. It is also reported that EM164 inhibits IGF-1, IGF-2, and serum-stimulated cell proliferation and survival of diverse human cancer cell lines in vitro, including breast, lung, colon, cervical, ovarian, pancreatic, melanoma, prostate, neuroblastoma, rhabdomyosarcoma, and osteosarcoma cancer lines [120, 121]. EM164 has shown clinical activity in malignant cancers and is currently being evaluated in patients with advanced metastatic cancer [122].
IMC-A14
IMC-A14 monoclonal antibodies have high affinity for the extracellular domain of IGF-1R and inhibit ligand binding with an IC50 of 0.6–1 nM. Ligand binding to the IGF-1R inhibited MAPK and PI3 K downstream signaling of IGF pathways, which ultimately led to the inhibition of mitogenic growth in addition to proliferative potential of IGF. IMC-A14 did not block the IR but might block binding to a typical IGF-1R variety of cancer cell lines [123].
IMC-A12
IMC-A12, another monoclonal antibody developed by Imclone systems Inc., generated by screening a human Fab phage display library, selectively binds to IGF-1R with IC50 of 0.6–1 nmol/L without effecting IR. The role of IGF in cell proliferation was antagonized by IMC-A12 via inhibiting mitogenic (MAPK) and metabolic (PI3 K) signaling pathways. Furthermore, xenograft tumor model experiment also demonstrated that IMC-A12 reduces the cell proliferation and induce apoptosis in breast, pancreatic, colon tumors, and many other neoplasms. Moreover, it is reported that most advanced therapy combination approach of IGF-1R-specific inhibitors and nonspecific chemotherapeutic agents or radiation have beneficial effect compared to conventional treatments alone. The phase I and II clinical trials of IMC-A12 demonstrated favorable, safe, and cost-effective therapeutic antibody in various cancers [124]. More interesting is the inhibition of IGF-1R brought about by IMC-A12 in both constitutive and IGF1-induced secretion of VEGF, indicating that a potent antiangiogenic mechanism was associated with the IMC-A1 treatment, which contributed to its antitumor effect [125]. These findings suggested that IMC-A12 is a therapeutic aspirant for both androgen-dependent and androgen-independent prostate cancer [126]. IMC-A12 markedly augmented the inhibition of docetaxel on tumor growth by the regulation of cell cycle progression and cell survival-associated genes [127].
Figitumumab (CP-751871)
CP-751871 is a monoclonal antibody targeting the IGF-1R. This had been studied in variety of cancers, including adrenocortical carcinoma, non-small cell lung cancer, and Ewing’s sarcoma [128–131]. Preclinical results demonstrate synergistic combination therapy of CP-751871 with mTOR inhibitor along with IGF-1R signaling are more beneficial and safe with no unexpected side effects [132]. A series of conventional drugs such as Temsirolimus were combined with cixutumumab, a fully human IgG1 monoclonal antibody intended to target IGF-1R resulted in antitumor activity against Ewing’s family tumors without side effects [133]. Recently, progress has been made toward the development of multitargeted agents targeting tyrosine kinase receptors along with other signaling pathways such as MAPK and PI3 K or combination of these agents, which can prove to be a beacon for the development of therapeutic options in a broad array of cancer tumors [134].
MK-0646
MK-0646, known as h7c10 or F50035, is a recombinant humanized IgG1 anti-IGF-1R monoclonal antibody reducing cell proliferation as well as inducing apoptosis in a dose-dependent manner. MK-0646 interrupts the IGF-1R-induced activation of PI3 K and MAPK pathways, which results in internalization and degradation of IGF-1R. This antibody is also seen to exhibit potent antitumor efficacy in ingrained tumor. However, combination therapy with other tyrosine kinase and anti-androgen chemotherapeutic agent may be more successful in the treatment for cancer than when used alone [135–137]. Moreover, it was found that, besides IGF-1R, this antibody recognizes and specifically binds to hybrid receptors. A phase I clinical trial study of MK-0646 in advanced solid tumor patients is in progress including pancreatic cancer, neuroendocrine tumors, and colorectal cancers [136, 138, 139].
19D12
19D12, fully humanized neutralizing anti-IGF-1R antibody, inhibits IGF-binding and autophosphorylation of both IGF-1R/IGF-1R homodimers and IGF-1R/IR heterodimers, but interestingly, 19D12 does not bind to the IR homodimers. In addition, to inhibiting IGF-1R autophosphorylation, 19D12 also inhibits the activation of the major downstream signaling molecules PI3 K, Akt, and mTOR that leads to the down-regulation of IGF-IR expression in vitro and in vivo [140]. In addition to inhibiting the in vitro proliferation of several cancer cell lines, this antibody proved fruitful in a various xenograft models, including ovarian (A2780), non-small cell lung cancer (H322), breast (MCF-7), and colon cancer (HT-29) [141–144].
ScFv-Fc-IGF-IR
Recombinant anti-IGF-IR antibody, scFv-Fc, consisting of 1H7 monoclonal antibody-derived single-chain antibody (scFv) and human IgG1 Fc extensively suppressed breast tumor growth [145–147]. Further, in vivo effects of this antibody on breast tumor growth in the absence or presence of tamoxifen on xenograft growth in athymic mice confirm the suppression of the tumor growth [148, 149].
Small single-chain peptide: block the binding of IR with ligands
Insulin action is evoked by binding of insulin to extracellular transmembrane α-subunits of its receptor, thereby engendering a conformational change in the receptor, which turn on the intracellular tyrosine kinase signaling cascade of β-subunits. Series of peptides, previously reported, forms covalent linkage to two separate hotspots on the IR, resulting in insulin agonistic or antagonistic functions, based on how the peptides are linked [150]. In the case of the IR, insulin dimer peptide has been shown to be antagonistic to the insulin receptor. However, synthetic small peptides targeting IR were shown to be either agonists or antagonists [151]. Thus, so far the only existing insulin antagonists have been IR antibodies [152]. A 43 amino acid single-chain small peptide, IR antagonist, S661 was developed by L Schäffer. The affinity of S661 for the IR was higher than that of insulin. S661 completely inhibits insulin action, both in cellular assays and in vivo in rats. Moreover, new peptide was synthesized by modification on S661 and this biosynthetic version was called S961, which was identical to S661 except for a C-terminal acid that was seen to have properties indistinguishable from those of S661. These antagonists are helpful for understanding the molecular mechanism of insulin receptor signaling and treatment for hypoglycemic conditions [153]. Further, in vivo results also suggested that S961 induces hyperglycemia, hyperinsulinemia, glucose intolerance in the insulin arbitrate glucose disposal in the Sprague Dawley rats [154].
Neutralization of IGF-1/IGF-2
IR and IGF-1R are activated by binding of its natural ligands. Neutralization of insulin, IGF-1, and IGF-2 ligands leads to the inhibition of the IR and IGF-1R receptors. IGFBP are a family of vertebrate proteins that owns the main function of regulating bioavailability of insulin, IGF-1 and IGF-2, to interact with the receptors. There are six types of IGFBP that serve as a carrier protein for IGF-1 that regulates cell activity in various ways [38]. The IGFBP family has six distinct subgroups, IGFBP1-6, categorized on the basis of conservation of gene organization, structural similarity, and binding affinity for IGFs. Across species, IGFBP-5 exhibits the highest sequence conservation while IGFBP-6 exhibits the least sequence conservation. A third method recently described to neutralize the ligand is the use of antibodies against IGF-1 and IGF-2 [33]. A rat monoclonal antibody, KM1468, which neutralized both IGF-1 and IGF-2, inhibited bone metastases of a variety of cancer cells, indicating that such an approach could have clinical value [155, 156].
Conclusions and future remarks
Most recent studies have suggested the concept that multitargeted receptor tyrosine kinase inhibitors are successful therapeutic targets for complex diseases like cancer and diabetes. Co-targeting the IRA and IGF-1R in cancer would be a more useful therapeutic option than targeting the IGF-1R alone. Preclinical data and early clinical trials have provided evidences that IRA/IGF-1R co-targeting may potentiate various chemotherapeutic regimens and prevent adaptive resistance to selective anti-IGF-1R drugs. Unfortunately, IR inhibition also deregulates the glucose metabolism. In this context, combinational drug acting as insulin sensitizers and IRA inhibitors, providing most beneficial options, is expected to be accessible in the near future. Co-targeting the mitogenic cascade of IRA along with IGF-1R may be most successful strategy for the treatment for complex diseases like cancer and diabetes in near future.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. Cancer J Clin. 2011;61:69–90.
Weiderpass E, Gridley G, Persson I, Nyrén O, Ekbom A, Adami HO. Risk of endometrial and breast cancer in patients with diabetes mellitus. Int J Cancer. 1997;71:360–3.
Wolf I, Sadetzki S, Catane R, Karasik A, Kaufman B. Diabetes mellitus and breast cancer. Lancet Oncol. 2005;6:103–11.
Bonovas S, Filioussi K, Tsantes A. Diabetes mellitus and risk of prostate cancer: a meta-analysis. Diabetologia. 2004;47:1071–8.
Kasper JS, Giovannucci E. A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prevent. 2006;15:2056–62.
Abreu-Martin MT, Chari A, Palladino AA, Craft NA, Sawyers CL. Mitogen-activated protein kinase kinase kinase 1 activates androgen receptor-dependent transcription and apoptosis in prostate cancer. Mol Cell Biol. 1999;19:5143–54.
Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr-Relat Cancer. 2009;16:1103–23.
Vigneri P, Frasca F, Sciacca L, Frittitta L, Vigneri R. Obesity and cancer. Nutr Metab Cardiovasc Dis. 2006;16:1–7.
Fisher WE. Diabetes: risk factor for the development of pancreatic cancer or manifestation of the disease? World J Surg. 2001;25:503–8.
Strickler HD, Wylie-Rosett J, Rohan T, Hoover DR, Smoller S, Burk RD, Yu H. The relation of type 2 diabetes and cancer. Diabetes Technol Ther. 2001;3:263–74.
Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–91.
Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, Rosner B, Speizer FE, Pollak M. Circulating concentrations of insulin-like growth factor I and risk of breast cancer. Lancet. 1998;351:1393–6.
Hsing AW, Sakoda LC, Chua SC. Obesity, metabolic syndrome, and prostate cancer. Am J Clin Nutr. 2007;86:843S–57S.
Tao Y, Pinzi V, Bourhis J, Deutsch E. Mechanisms of disease: signaling of the insulin-like growth factor 1 receptor pathway—therapeutic perspectives in cancer. Nat Rev Clin Oncol. 2007;4:591–602.
Djavan B, Waldert M, Seitz C, Marberger M. Insulin-like growth factors and prostate cancer. World J Urol. 2001;19:225–33.
Gualberto A, Pollak M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinical trial results and future directions. Oncogene. 2009;28:3009–21.
Rodon J, DeSantos V, Ferry RJ, Kurzrock R. Early drug development of inhibitors of the insulin-like growth factor-I receptor pathway: lessons from the first clinical trials. Mol Cancer Ther. 2008;7:2575–88.
Zhang H, Fagan DH, Zeng X, Freeman KT, Sachdev D, Yee D. Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene. 2010;29:2517–27.
Ryo M, Nakamura T, Kihara S, Kumada M, Shibazaki S, Takahashi M, Nagai M, Matsuzawa Y, Funahashi T. Adiponectin as a biomarker of the metabolic syndrome. Circ J. 2004;68:975.
Grundy M, Scott M. Hypertriglyceridemia, atherogenic dyslipidemia, and the metabolic syndrome. Am J Cardiol. 1998;81:18B–25B.
Nguyen NT, Magno CP, Lane KT, Hinojosa MW, Lane JS. Association of hypertension, diabetes, dyslipidemia, and metabolic syndrome with obesity: findings from the National Health and Nutrition Examination Survey, 1999 to 2004. J Am Coll Surg. 2008;207:928–34.
Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–18.
Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–28.
Zhang H, Yee D. The therapeutic potential of agents targeting the type I insulin-like growth factor receptor. Expert Opin Inv Drugs. 2004;13:1569–77.
Hofmann F, García-Echeverría C. Blocking the insulin-like growth factor-I receptor as a strategy for targeting cancer. Drug Discov Today. 2005;10:1041–8.
Gennigens C, Menetrier-Caux C, Droz J. Insulin-like growth factor (IGF) family and prostate cancer. Crit Rev Oncol Hematol. 2006;58:124.
Huang J, Morehouse C, Streicher K, Higgs BW, Gao J, Czapiga M, Boutrin A, Zhu W, Brohawn P, Chang Y. Altered expression of insulin receptor isoforms in breast cancer. PLoS ONE. 2011;6:e26177.
Heni M, Hennenlotter J, Scharpf M, Lutz SZ, Schwentner C, Todenhöfer T, Schilling D, Kühs U, Gerber V, Machicao F. Insulin receptor isoforms A and B as well as insulin receptor substrates-1 and 2 are differentially expressed in prostate cancer. PLoS ONE. 2012;7:e50953.
Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–69.
Malaguarnera R, Sacco A, Voci C, Pandini G, Vigneri R, Belfiore A. Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology. 2012;153:2152–63.
Dupont J, Holzenberger M. Biology of insulin like growth factors in development. Birth Defects Res C Embryo Today. 2003;69:257–71.
Xu G, Marshall CA, Lin TA, Kwon G, Munivenkatappa RB, Hill JR, Lawrence JC. McDaniel ML Insulin mediates glucose-stimulated phosphorylation of PHAS-I by pancreatic beta cells. J Biol Chem. 1998;273:4485.
Sachdev D, Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007;6:1–12.
Duan C, Xu Q. Roles of insulin-like growth factor (IGF) binding proteins in regulating IGF actions. Gen Comp Endocrinol. 2005;142:44–52.
De Meyts P, Whittaker J. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov. 2002;1:769–83.
Beauchamp M-C, Yasmeen A, Knafo A, Gotlieb WH. Targeting insulin and insulin-like growth factor pathways in epithelial ovarian cancer. J Oncol. 2010. doi:10.1155/2010/257058.
LeRoith D, Roberts CT. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–37.
Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocrine Rev. 1999;20:761–87.
Baxter RC. Insulin-like growth factor binding proteins in the human circulation: a review. Horm Res Paediatr. 1994;42:140–4.
Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet. 2004;363:1346–53.
Jerome L, Alami N, Belanger S, Page V, Yu Q, Paterson J, Shiry L, Pegram M, Leyland-Jones B. Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2-overexpressing breast tumors and potentiates Herceptin activity in vivo. Cancer Res. 2006;66:7245–52.
Nahta R, Yu D, Hung M-C, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Rev Clin Oncol. 2006;3:269–80.
Fuller GN, Rhee CH, Hess KR, Caskey LS, Wang R, Bruner JM, Yung WA, Zhang W. Reactivation of insulin-like growth factor binding protein 2 expression in glioblastoma multiforme a revelation by parallel gene expression profiling. Cancer Res. 1999;59:4228–32.
Busund L, Richardsen E, Busund R, Ukkonen T, Bjørnsen T, Busch C, Stalsberg H. Significant expression of IGFBP2 in breast cancer compared with benign lesions. J Clin Pathol. 2005;58:361–6.
Wang H, Wang H, Shen W, Huang H, Hu L, Ramdas L, Zhou Y-H, Liao WS, Fuller GN, Zhang W. Insulin-like growth factor binding protein 2 enhances glioblastoma invasion by activating invasion-enhancing genes. Cancer Res. 2003;63:4315–21.
Dupont J, LeRoith D. Insulin and insulin-like growth factor I receptors: similarities and differences in signal transduction. Horm Res Paediatr. 2000;55:22–6.
Frasca F, Pandini G, Sciacca L, Pezzino V, Squatrito S, Belfiore A, Vigneri R. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch Physiol Biochem. 2008;114:23–37.
Kuijjer ML, Peterse EF, van den Akker BE, Briaire-de Bruijn IH, Serra M, Meza-Zepeda LA, Myklebost O, Hassan AB, Hogendoorn PC, Cleton-Jansen A-M. IR/IGF1R signaling as potential target for treatment of high-grade osteosarcoma. BMC Cancer. 2013;13:245.
Vella V, Sciacca L, Pandini G, Mineo R, Squatrito S, Vigneri R, Belfiore A. The IGF system in thyroid cancer: new concepts. Mol Pathol. 2001;54:121–4.
Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, Siddle K, Goldfine ID, Belfiore A. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res. 1999;5:1935–44.
Cox ME, Gleave ME, Zakikhani M, Bell RH, Piura E, Vickers E, Cunningham M, Larsson O, Fazli L, Pollak M. Insulin receptor expression by human prostate cancers. Prostate. 2009;69:33–40.
Werner H, Weinstein D, Bentov I. Similarities and differences between insulin and IGF-I: structures, receptors, and signalling pathways. Arch Physiol Biochem. 2008;114:17–22.
Kissau L, Stahl P, Mazitschek R, Giannis A, Waldmann H. Development of natural product-derived receptor tyrosine kinase inhibitors based on conservation of protein domain fold. J Med Chem. 2003;46:2917–31.
Denley A, Bonython ER, Booker GW, Cosgrove LJ, Forbes BE, Ward CW, Wallace JC. Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol Endocrinol. 2004;18:2502–12.
Denley A, Cosgrove LJ, Booker GW, Wallace JC, Forbes BE. Molecular interactions of the IGF system. Cytokine Growth Factor Rev. 2005;16:421–39.
Zhang H, Pelzer AM, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res. 2007;67:391–7.
Pierre-Eugene C, Pagesy P, Nguyen TT, Neuillé M, Tschank G, Tennagels N, Hampe C. Issad T Effect of insulin analogues on insulin/IGF1 hybrid receptors: increased activation by glargine but not by its metabolites M1 and M2. PLoS ONE. 2012;7:e41992.
Blanquart C, Achi J, Issad T. Characterization of IRA/IRB hybrid insulin receptors using bioluminescence resonance energy transfer. Biochem Pharmacol. 2008;76:873–83.
Sherajee SJ, Fujita Y, Rafiq K, Nakano D, Mori H, Masaki T, Hara T, Kohno M, Nishiyama A, Hitomi H. Aldosterone induces vascular insulin resistance by increasing insulin-like growth factor-1 receptor and hybrid receptor. Arterioscler Thromb Vasc Biol. 2012;32:257–63.
Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002;277:39684–95.
Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocrine Rev. 2009;30:586.
Mamula PW, McDonald AR, Brunetti A, Okabayashi Y, Wong KY, Maddux BA, Logsdon C, Goldfine ID. Regulating insulin-receptor-gene expression by differentiation and hormones. Diabetes Care. 1990;13:288–301.
Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem. 2008;114:63–70.
Wittman M, Carboni J, Attar R, Balasubramanian B, Balimane P, Brassil P, Beaulieu F, Chang C, Clarke W, Dell J. Discovery of a 1 H-Benzoimidazol-2-yl)-1 H-pyridin-2-one (BMS-536924) inhibitor of insulin-like growth factor I receptor kinase with in vivo antitumor activity. J Med Chem. 2005;48:5639–43.
Beauchamp M-C, Knafo A, Yasmeen A, Carboni JM, Gottardis MM, Pollak MN, Gotlieb WH. BMS-536924 sensitizes human epithelial ovarian cancer cells to the PARP inhibitor, 3-aminobenzamide. Gynecol Oncol. 2009;115:193–8.
Velaparthi U, Saulnier MG, Wittman MD, Liu P, Frennesson DB, Zimmermann K, Carboni JM, Gottardis M, Li A, Greer A. Insulin-like growth factor-1 receptor (IGF-1R) kinase inhibitors: SAR of a series of 3-[6-(4-substituted-piperazin-1-yl)-4-methyl-1H-benzimidazol-2-yl]-1H-pyridine-2-one. Bioorg Med Chem Lett. 2010;20:3182–5.
Avnet S, Sciacca L, Salerno M, Gancitano G, Cassarino MF, Longhi A, Zakikhani M, Carboni JM, Gottardis M, Giunti A. Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma. Cancer Res. 2009;69:2443–52.
Hendrickson AEW, Haluska P, Schneider PA, Loegering DA, Peterson KL, Attar R, Smith BD, Erlichman C, Gottardis M, Karp JE. Expression of insulin receptor isoform A and insulin-like growth factor-1 receptor in human acute myelogenous leukemia: effect of the dual-receptor inhibitor BMS-536924 in vitro. Cancer Res. 2009;69:7635–43.
Dool CJ, Mashhedi H, Zakikhani M, David S, Zhao Y, Birman E, Carboni JM, Gottardis M, Blouin M-J, Pollak M. IGF1/insulin receptor kinase inhibition by BMS-536924 is better tolerated than alloxan-induced hypoinsulinemia and more effective than metformin in the treatment of experimental insulin-responsive breast cancer. Endocr Relat Cancer. 2011;18:699–709.
Huang F, Greer A, Hurlburt W, Han X, Hafezi R, Wittenberg GM, Reeves K, Chen J, Robinson D, Li A. The mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. Cancer Res. 2009;69:161–70.
Jin Q, Esteva FJ. Cross-talk between the ErbB/HER family and the type I insulin-like growth factor receptor signaling pathway in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:485–98.
Litzenburger BC, Creighton CJ, Tsimelzon A, Chan BT, Hilsenbeck SG, Wang T, Carboni JM, Gottardis MM, Huang F, Chang JC. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin Cancer Res. 2011;17:2314–27.
Wittman MD, Carboni JM, Yang Z, Lee FY, Antman M, Attar R, Balimane P, Chang C, Chen C, Discenza L. Discovery of a 2,4-disubstituted pyrrolo [1,2-f][1,2,4] triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. J Med Chem. 2009;52:7360–3.
Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther. 2009;8:3341–9.
Huang F, Hurlburt W, Greer A, Reeves KA, Hillerman S, Chang H, Fargnoli J, Finckenstein FG, Gottardis MM, Carboni JM. Differential mechanisms of acquired resistance to insulin-like growth factor-I receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010;70:7221–31.
Hou X, Huang F, Macedo LF, Harrington SC, Reeves KA, Greer A, Finckenstein FG, Brodie A, Gottardis MM, Carboni JM. Dual IGF-1R/InsR inhibitor BMS-754807 synergizes with hormonal agents in treatment of estrogen-dependent breast cancer. Cancer Res. 2011;71:7597–607.
Dinchuk JE, Cao C, Huang F, Reeves KA, Wang J, Myers F, Cantor GH, Zhou X, Attar RM, Gottardis M. Insulin receptor (IR) pathway hyperactivity in IGF-IR null cells and suppression of downstream growth signaling using the dual IGF-IR/IR inhibitor, BMS-754807. Endocrinology. 2010;151:4123–32.
Kolb EA, Gorlick R, Lock R, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH, Maris JM, Billups C. Initial testing (stage 1) of the IGF-1 receptor inhibitor BMS-754807 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2011;56:595–603.
Desai J, Solomon B, Davis I, Lipton L, Hicks R, Scott A, Park J, Clemens P, Gestone T, Finckenstein F. Phase I dose-escalation study of daily BMS-754807, an oral, dual IGF-1R/insulin receptor (IR) inhibitor in subjects with solid tumors. J Clin Oncol. 2010;28:15s, abstr 3104.
Levitzki A, Mishani E. Tyrphostins and other tyrosine kinase inhibitors. Annu Rev Biochem. 2006;75:93–109.
Gazit A, Osherov N, Posner I, Yaish P, Poradosu E, Gilon C, Levitzki A. Tyrphostins. II. Heterocyclic and alpha-substituted benzylidenemalononitrile tyrphostins as potent inhibitors of EGF receptor and ErbB2/neu tyrosine kinases. J Med Chem. 1991;34:1896–907.
Davoodi-Semiromi A, Wasserfall CH, Xia CQ, Cooper-DeHoff RM, Wabitsch M, Clare-Salzler M, Atkinson M. The tyrphostin agent AG490 prevents and reverses type 1 diabetes in NOD mice. PLoS ONE. 2012;7:e36079.
Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, O’Connor M, Pirritt C, Sun Y, Yao Y. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem. 2009;1:1153–71.
King ER, Wong K–K. Insulin-like growth factor: current concepts and new developments in cancer therapy. Recent Pat Anti-Cancer Drug Discov. 2012;7:14–30.
Pitts TM, Tan AC, Kulikowski GN, Tentler JJ, Brown AM, Flanigan SA, Leong S, Coldren CD, Hirsch FR, Varella-Garcia M. Development of an integrated genomic classifier for a novel agent in colorectal cancer: approach to individualized therapy in early development. Clin Cancer Res. 2010;16:3193–204.
Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, Lerner L, Chiu MI, Wild R, Epstein D. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther. 2010;9:2652–64.
Zeng X, Zhang H, Oh A, Zhang Y, Yee D. Enhancement of doxorubicin cytotoxicity of human cancer cells by tyrosine kinase inhibition of insulin receptor and type I IGF receptor. Breast Cancer Res Treat. 2012;133:117–26.
King ER, Zu Z, Tsang Y, Deavers MT, Malpica A, Mok SC, Gershenson DM, Wong K–K. The insulin-like growth factor 1 pathway is a potential therapeutic target for low-grade serous ovarian carcinoma. Gynecol Oncol. 2011;123:13–8.
McKinley ET, Bugaj JE, Zhao P, Guleryuz S, Mantis C, Gokhale PC, Wild R, Manning HC. 18FDG-PET predicts pharmacodynamic response to OSI-906, a dual IGF-1R/IR inhibitor, in preclinical mouse models of lung cancer. Clin Cancer Res. 2011;17:3332–40.
Flanigan SA, Pitts TM, Eckhardt SG, Tentler JJ, Tan AC, Thorburn A, Leong S. The insulin-like growth factor I receptor/insulin receptor tyrosine kinase inhibitor PQIP exhibits enhanced antitumor effects in combination with chemotherapy against colorectal cancer models. Clin Cancer Res. 2010;16:5436–46.
Haluska P, Carboni JM, Loegering DA, Lee FY, Wittman M, Saulnier MG, Frennesson DB, Kalli KR, Conover CA, Attar RM. In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Res. 2006;66:362–71.
Ekman S, Frödin J-E, Harmenberg J, Bergman A, Hedlund Å, Dahg P, Alvfors C, Ståhl B, Bergström S, Bergqvist M. Clinical phase I study with an insulin-like growth factor-1 receptor inhibitor: experiences in patients with squamous non-small cell lung carcinoma. Acta Oncol. 2011;50:441–7.
Scagliotti GV, Novello S. The role of the insulin-like growth factor signaling pathway in non-small cell lung cancer and other solid tumors. Cancer Treat Rev. 2012;38:292–302.
Girnita A, All-Ericsson C, Economou MA, Axelson M, Seregard S, Larsson O, Girnita L. The insulin-like growth factor-I receptor inhibitor picropodophyllin causes tumor regression and attenuates mechanisms involved in invasion of uveal melanoma cells. Clin Cancer Res. 2006;12:1383–91.
Menu E, Jernberg-Wiklund H, Stromberg T, De Raeve H, Girnita L, Larsson O, Axelson M, Asosingh K, Nilsson K, Van Camp B. Inhibiting the IGF-1 receptor tyrosine kinase with the cyclolignan PPP: an in vitro and in vivo study in the 5T33MM mouse model. Blood. 2006;107:655–60.
Feng X, Aleem E, Lin Y, Axelson M, Larsson O, Strömberg T. Multiple antitumor effects of picropodophyllin in colon carcinoma cell lines: clinical implications. Int J Oncol. 2012;40:1251.
García-Echeverría C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro H-G, Cozens R. In vivo antitumor activity of NVP-AEW541, a novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231–9.
Scotlandi K, Manara MC, Nicoletti G, Lollini P-L, Lukas S, Benini S, Croci S, Perdichizzi S, Zambelli D, Serra M. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res. 2005;65:3868–76.
Tanno B, Mancini C, Vitali R, Mancuso M, McDowell HP, Dominici C, Raschellà G. Down-regulation of insulin-like growth factor I receptor activity by NVP-AEW541 has an antitumor effect on neuroblastoma cells in vitro and in vivo. Clin Cancer Res. 2006;12:6772–80.
Tazzari P, Tabellini G, Bortul R, Papa V, Evangelisti C, Grafone T, Martinelli G, McCubrey J, Martelli A. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia. 2007;21:886–96.
Mukohara T, Shimada H, Ogasawara N, Wanikawa R, Shimomura M, Nakatsura T, Ishii G, Park JO, Jänne PA, Saijo N. Sensitivity of breast cancer cell lines to the novel insulin-like growth factor-1 receptor (IGF-1R) inhibitor NVP-AEW541 is dependent on the level of IRS-1 expression. Cancer Lett. 2009;282:14–24.
Maiso P, Ocio EM, Garayoa M, Montero JC, Hofmann F, García-Echeverría C, Zimmermann J, Pandiella A, San Miguel JF. The insulin-like growth factor-I receptor inhibitor NVP-AEW541 provokes cell cycle arrest and apoptosis in multiple myeloma cells. Brit J Haematol. 2008;141:470–82.
Gariboldi MB, Ravizza R, Monti E. The IGFR1 inhibitor NVP-AEW541 disrupts a pro-survival and pro-angiogenic IGF-STAT3-HIF1 pathway in human glioblastoma cells. Biochem Pharmacol. 2010;80:455–62.
Baumann P, Hagemeier H, Mandl-Weber S, Franke D, Schmidmaier R. Myeloma cell growth inhibition is augmented by synchronous inhibition of the insulin-like growth factor-1 receptor by NVP-AEW541 and inhibition of mammalian target of rapamycin by Rad001. Anticancer Drugs. 2009;20:259–66.
Moser C, Schachtschneider P, Lang SA, Gaumann A, Mori A, Zimmermann J, Schlitt HJ, Geissler EK, Stoeltzing O. Inhibition of insulin-like growth factor-I receptor (IGF-IR) using NVP-AEW541, a small molecule kinase inhibitor, reduces orthotopic pancreatic cancer growth and angiogenesis. Eur J Cancer. 2008;44:1577–86.
Wolf S, Lorenz J, Mössner J, Wiedmann M. Treatment of biliary tract cancer with NVP-AEW541: mechanisms of action and resistance. World J Gastroenterol. 2010;16:156.
Tarn C, Rink L, Merkel E, Flieder D, Pathak H, Koumbi D, Testa JR, Eisenberg B, von Mehren M, Godwin AK. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci USA. 2008;105:8387–92.
Manara MC, Landuzzi L, Nanni P, Nicoletti G, Zambelli D, Lollini PL, Nanni C, Hofmann F, García-Echeverría C, Picci P. Preclinical in vivo study of new insulin-like growth factor-I receptor-specific inhibitor in Ewing’s sarcoma. Clin Cancer Res. 2007;13:1322–30.
Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, Giordano TJ, Ben-Josef E, Hammer GD. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab. 2009;94:204–10.
Gotlieb WH, Bruchim I, Gu J, Shi Y, Camirand A, Blouin M-J, Zhao Y, Pollak MN. Insulin-like growth factor receptor I targeting in epithelial ovarian cancer. Gynecol Oncol. 2006;100:389–96.
Hägerstrand D, Lindh MB, Peña C, Garcia-Echeverria C, Nistér M, Hofmann F, Östman A. PI3 K/PTEN/Akt pathway status affects the sensitivity of high-grade glioma cell cultures to the insulin-like growth factor-1 receptor inhibitor NVP-AEW541. Neuro-Oncology. 2010;12:967–75.
Martins AS, Mackintosh C, Martín DH, Campos M, Hernández T, Ordóñez J-L, de Alava E. Insulin-like growth factor I receptor pathway inhibition by ADW742, alone or in combination with imatinib, doxorubicin, or vincristine, is a novel therapeutic approach in Ewing tumor. Clin Cancer Res. 2006;12:3532–40.
Scotlandi K, Manara MC, Hattinger CM, Benini S, Perdichizzi S, Pasello M, Bacci G, Zanella L, Bertoni F, Picci P. Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing’s sarcoma. Eur J Cancer. 2005;41:1349–61.
Lu D, Jimenez X, Zhang H, Atkins A, Brennan L, Balderes P, Bohlen P, Witte L, Zhu Z. Di-diabody: a novel tetravalent bispecific antibody molecule by design. J Immunol Methods. 2003;279:219–32.
Kontermann RE. Recombinant bispecific antibodies for cancer therapy. Acta Pharmacol Sinica. 2005;26:1–9.
Deyev SM, Lebedenko EN. Multivalency: the hallmark of antibodies used for optimization of tumor targeting by design. BioEssays. 2008;30:904–18.
Hartog H, Wesseling J, Boezen HM, van der Graaf WT. The insulin-like growth factor 1 receptor in cancer: old focus, new future. Eur J Cancer. 2007;43:1895–904.
Ozkan EE. Plasma and tissue insulin-like growth factor-I receptor (IGF-IR) as a prognostic marker for prostate cancer and anti-IGF-IR agents as novel therapeutic strategy for refractory cases: a review. Mol Cell Endocrinol. 2011;344:1–24.
Hewish M, Chau I, Cunningham D. Insulin-like growth factor 1 receptor targeted therapeutics: novel compounds and novel treatment strategies for cancer medicine. Recent Pat Anticancer Drug Discov. 2009;4:54–72.
Maloney EK, McLaughlin JL, Dagdigian NE, Garrett LM, Connors KM, Zhou X-M, Blättler WA, Chittenden T, Singh R. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res. 2003;63:5073–83.
Zeng X, Sachdev D, Zhang H, Gaillard-Kelly M, Yee D. Sequencing of type I insulin-like growth factor receptor inhibition affects chemotherapy response in vitro and in vivo. Clin Cancer Res. 2009;15:2840–9.
Geoerger B, Brasme J-F, Daudigeos-Dubus E, Opolon P, Venot C, Debussche L, Vrignaud P, Vassal G. Anti-insulin-like growth factor 1 receptor antibody EM164 (murine AVE1642) exhibits anti-tumour activity alone and in combination with temozolomide against neuroblastoma. Eur J Cancer. 2010;46:3251–62.
Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, Bassi R, Abdullah R, Hooper AT, Koo H. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res. 2003;63:8912–21.
Rowinsky EK, Youssoufian H, Tonra JR, Solomon P, Burtrum D, Ludwig DL. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin Cancer Res. 2007;13:5549–55.
Wu K-D, Zhou L, Burtrum D, Ludwig DL, Moore MA. Antibody targeting of the insulin-like growth factor I receptor enhances the anti-tumor response of multiple myeloma to chemotherapy through inhibition of tumor proliferation and angiogenesis. Cancer Immunol Immunother. 2007;56:343–57.
Wu JD, Odman A, Higgins LM, Haugk K, Vessella R, Ludwig DL, Plymate SR. In vivo effects of the human type I insulin-like growth factor receptor antibody A12 on androgen-dependent and androgen-independent xenograft human prostate tumors. Clin Cancer Res. 2005;11:3065–74.
Wu JD, Haugk K, Coleman I, Woodke L, Vessella R, Nelson P, Montgomery RB, Ludwig DL, Plymate SR. Combined in vivo effect of A12, a type 1 insulin-like growth factor receptor antibody, and docetaxel against prostate cancer tumors. Clin Cancer Res. 2006;12:6153–60.
Haluska P, Worden F, Olmos D, Yin D, Schteingart D, Batzel GN, Paccagnella ML, de Bono JS, Gualberto A, Hammer GD. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother Pharmacol. 2010;65:765–73.
Gualberto A, Karp DD. Development of the monoclonal antibody Figitumumab, targeting the insulin-like growth factor-1 receptor, for the treatment of patients with non-small-cell lung cancer. Clin Lung Cancer. 2009;10:273–80.
Olmos D, Postel-Vinay S, Molife L, Okuno SH, Schuetze SM, Paccagnella ML, Batzel GN, Yin D, Pritchard-Jones K, Judson I. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 2010;11:129–35.
O’Neill A, Shah N, Zitomersky N, Ladanyi M, Shukla N, Üren A, Loeb D, Toretsky J. Insulin-like growth factor 1 receptor as a therapeutic target in Ewing Sarcoma: lack of consistent upregulation or recurrent mutation and a review of the clinical trial literature. Sarcoma. 2013.
Quek R, Wang Q, Morgan JA, Shapiro GI, Butrynski JE, Ramaiya N, Huftalen T, Jederlinic N, Manola J, Wagner AJ. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res. 2011;17:871–9.
Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, Ludwig J, Chen HX, Doyle LA, Kurzrock R. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors. Clin Cancer Res. 2012;18:2625–31.
Hixon ML, Paccagnella L, Millham R, Perez-Olle R, Gualberto A. Development of inhibitors of the IGF-IR/PI3 K/Akt/mTOR pathway. Rev Recent Clin Trials. 2010;5:189–208.
Javle M, Varadhachary G, Bhosale P, Ukegbu L, Overman M, Shroff R. Phase I study of MK-0646, a humanized monoclonal antibody against IGF-1R in combination with gemcitabine or gemcitabine plus erlotinib (E) for advanced previously untreated pancreatic cancer. ASCO gastrointestinal cancers symposium (abstract 131), 2010.
Watkins D, Tabernero J, Schmoll H, Trarbach T, Ramos F, Hsu K, Gates M, Clark J, LeVan P, Cunningham D. A phase II study of the anti-IGFR antibody MK-0646 in combination with cetuximab and irinotecan in the treatment of chemorefractory metastatic colorectal cancer. J Clin Oncol. 2009;27:15s, abstr 4127.
Bao XH, Naomoto Y, Hao HF, Watanabe N, Sakurama K, Noma K, Motoki T, Tomono Y, Fukazawa T, Shirakawa Y. IGF-IR and its inhibitors in gastrointestinal carcinomas (Review). Oncol Lett. 2010;1:195–201.
Atzori F, Tabernero J, Cervantes A, Prudkin L, Andreu J, Rodríguez-Braun E, Domingo A, Guijarro J, Gamez C, Rodon J. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011;17:6304–12.
Javle M, Varadhachary G, Shroff R, Bhosale P, Overman M, Weatherly J, Wolff R, Abbruzzese J. Phase I/II study of MK-0646, the humanized monoclonal IGF-1R antibody in combination with gemcitabine or gemcitabine plus erlotinib (E) for advanced pancreatic cancer. J Clin Oncol. 2010; meeting abstr 4039.
Wang Y, Hailey J, Williams D, Wang Y, Lipari P, Malkowski M, Wang X, Xie L, Li G, Saha D. Inhibition of insulin-like growth factor-I receptor (IGF-IR) signaling and tumor cell growth by a fully human neutralizing anti-IGF-IR antibody. Mol Cancer Ther. 2005;4:1214–21.
Wang Y, Ji Q-s, Mulvihill M, Pachter JA. Inhibition of the IGF-I receptor for treatment of cancer. Kinase inhibitors and monoclonal antibodies as alternative approaches. In: Targeted interference with signal transduction events. Recent results in cancer research, vol. 17, 2007. p. 59–76.
Kolb EA, Gorlick R, Houghton PJ, Morton CL, Lock R, Carol H, Reynolds CP, Maris JM, Keir ST, Billups CA. Initial testing (stage 1) of a monoclonal antibody (SCH 717454) against the IGF-1 receptor by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1190–7.
Wang X, Lipari P, Liu L, Long B, Liu J, Ramos R, Hailey J, Mayer-Ezell R, Wang L, Maxwell E. Efficacy of human anti-IGF-1R antibody in tumor xenograft models as a single agent and in combination with anti-cancer drugs. AACR Meeting Abstracts, 2005, p. 1190.
Wang Y, Hailey J, Williams D, Wang Y, Lipari P, Malkowski M, Wang X, Xie L, Li G, Saha D. Inhibition of IGF-1R signaling and tumor cell proliferation by a fully human neutralizing anti-IGF-1R antibody. In: Proceedings of the American Association for Cancer Research 2005:1190.
Ohtani M, Numazaki M, Yajima Y, Fujita-Yamaguchi Y. Mechanisms of antibody-mediated insulin-like growth factor I receptor (IGF-IR) down-regulation in MCF-7 breast cancer cells. Biosci Trends. 2009;3:131.
Li S-L, Liang S-J, Guo N, Wu AM, Fujita-Yamaguchi Y. Single-chain antibodies against human insulin-like growth factor I receptor: expression, purification, and effect on tumor growth. Cancer Immunol Immunother. 2000;49:243–52.
Fujita-Yamaguchi Y. Single-chain antibodies against human insulin-like growth factor I receptor: expression, purification, and effect on tumor growth. In: Google Patents. 2002.
Ye J–J, Liang S-J, Guo N, Li S-L, Wu A, Giannini S, Sachdev D, Yee D, Brünner N, Ikle D. Combined effects of tamoxifen and a chimeric humanized single chain antibody against the type I IGF receptor on breast tumor growth in vivo. Horm Metab Res. 2003;35:836–42.
Sachdev D, Li S-L, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D. A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res. 2003;63:627–35.
Schäffer L, Brissette RE, Spetzler JC, Pillutla RC, Østergaard S, Lennick M, Brandt J, Fletcher PW, Danielsen GM, Hsiao K-C. Assembly of high-affinity insulin receptor agonists and antagonists from peptide building blocks. Proc Natl Acad Sci USA. 2003;100:4435–9.
Knudsen L, Hansen BF, Jensen P, Pedersen TÅ, Vestergaard K, Schäffer L, Blagoev B, Oleksiewicz MB, Kiselyov VV, De Meyts P. Agonism and antagonism at the insulin receptor. PLoS ONE. 2012;7:e51972.
Soos M, Siddle K, Baron MD, Heward JM, Luzio JP, Bellatin J, Lennox ES. Monoclonal antibodies reacting with multiple epitopes on the human insulin receptor. Biochem J. 1986;235:199–208.
Schäffer L, Brand CL, Hansen BF, Ribel U, Shaw AC, Slaaby R, Sturis J. A novel high-affinity peptide antagonist to the insulin receptor. Biochem Biophys Res Commun. 2008;376:380–3.
Vikram A, Jena G. S961, an insulin receptor antagonist causes hyperinsulinemia, insulin-resistance and depletion of energy stores in rats. Biochem Biophys Res Commun. 2010;398:260–5.
Miyamoto S, Nakamura M, Shitara K, Nakamura K, Ohki Y, Ishii G, Goya M, Kodama K, Sangai T, Maeda H. Blockade of paracrine supply of insulin-like growth factors using neutralizing antibodies suppresses the liver metastasis of human colorectal cancers. Clin Cancer Res. 2005;11:3494–502.
Goya M, Miyamoto S, Nagai K, Ohki Y, Nakamura K, Shitara K, Maeda H, Sangai T, Kodama K, Endoh Y. Growth inhibition of human prostate cancer cells in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice by a ligand-specific antibody to human insulin-like growth factors. Cancer Res. 2004;64:6252–8.
Acknowledgments
We would like to thank Vice Chancellor, Central University of Punjab, Bathinda, Punjab, (India) for supporting this study with infrastructural requirements. We also thank Professor P. Ramarao (Dean, Academic Affairs) Central University of Punjab, Bathinda, Punjab, India, for his suggestions during the course that tremendously helped to improve this article. This study was also supported by a Senior Research Fellowship Grant-in-Aid from Indian Council of Medical Research (ICMR), Government of India awarded to PS.
Conflict of interest
The authors declare that there is no conflict of interests regarding the publication of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Singh, P., Alex, J.M. & Bast, F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol 31, 805 (2014). https://doi.org/10.1007/s12032-013-0805-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-013-0805-3