
Abstract
Background
Intestinal fibrosis is a common complication of Crohn’s disease (CD). Its exact mechanism is still unclear, and effective treatments to control or reverse the fibrosis process are unavailable. Epithelial–mesenchymal transition (EMT) may promote intestinal fibrosis by increasing deposition of extracellular matrix protein. IL-17A is a pro-inflammatory cytokine, and it has been shown as a profibrotic factor as its association with fibrosis of multiple organs was reported.
Aims
To assess the roles of IL-17A and EMT in the initiation and development of intestinal fibrosis and to verify the potential inductive effect of IL-17A on EMT.
Methods
In this study, we evaluated the expression of IL-17A and EMT-related genes in colonic mucosal biopsy tissues of CD patients and control individuals. Then, we examined the changes of EMT-related genes and fibrosis-related genes of IEC-6 cells which cultured for 72 h under increasing concentrations of IL-17A or with TGF-β1, to verify the potential inductive effect of IL-17A on EMT in vitro. We blocked the IL-17A of the mouse model of TNBS-induced experimental intestinal colitis and fibrosis to further verify the potential inductive effect of IL-17A on EMT in vivo.
Results
We found the occurrence of EMT and high-level expression of IL-17A in intestinal mucosa of CD patients. Using IEC-6 cells, we showed that IL-17A may induce EMT in intestinal epithelial cells that come with reduced E-cadherin expression and increased expression of vimentin, snail, and α-SMA. We further found that anti-IL-17A treatment alleviated intestinal fibrosis through reducing EMT in mouse intestine.
Conclusions
Our study confirmed the involvement of IL-17A in the development of intestinal fibrosis through inducing EMT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crohn’s disease (CD) is a chronic nonspecific intestinal disease of unknown etiology and pathogenesis. It is characterized by chronic relapses and may lead to fibrosis, intestinal stricture, and even obstruction [1]. Although many new drugs (such as biologics) have been developed to control inflammation, little is achieved in prevention of intestinal fibrosis [2, 3]; many CD patients will need surgical treatment for fibrotic [4]. Intestinal strictures occur in about one-third of CD patients [5], among those two-third of them received at least one surgical treatment within 20 years after diagnosis [6]. Such treatment suffers from poor long-term effectiveness, high rate of relapse, and high economical and psychological burden upon patients [7]. Therefore, it is of great significance to discover the molecular mechanisms of intestinal fibrosis so as to develop effective therapeutic strategies that could slow down or block the fibrotic process.
Development of intestinal fibrosis shares common mechanisms with the development of fibrosis in other organs [8]. Extracellular matrix (ECM) is composed mainly of macromolecules produced by mesenchymal cells which can directly or indirectly promote intestinal fibrosis [9]. Intestinal mesenchymal cells include fibroblasts, myofibroblasts, and smooth muscle cells [10]. Activation and proliferation of mesenchymal cells are important mechanisms in intestinal fibrotic process [8, 10]. Mesenchymal cells are activated by autocrine factors, paracrine signals, and pathogen-associated molecular patterns (PAMPs) [11, 12]. Proliferation of mesenchymal cells may derive from a variety of sources including dedifferentiation of epithelial cells by EMT [13,14,15] and endothelial cells via epithelial/endothelial–mesenchymal transition (EndoMT) [16], stellate cells [17], circulating precursors [18], and bone marrow stem cells [19].
While EMT plays important roles in pathological processes such as organ fibrosis and tumor progression [20, 21], during EMT, epithelial cells lose their original epithelial hallmarks including apico-basal polarization and specific intercellular junctions while adopting mesenchymal cell features. Characteristics of EMT include reduced expression of specific epithelial proteins such as E-cadherin or claudin-4, together with increased expression of mesenchymal-specific proteins such as vimentin, alpha smooth muscle actin (α-SMA), or fibroblast-specific protein 1 (FSP1). This is partially due to coordinated regulation of a distinct set of transcription factors, such as SNAIL1, SLUG, or TWIST [22]. Uncontrolled continuous transition from epithelial cells to myofibroblasts may lead to fibrosis, and there has been evidence suggesting EMT is the key pathological mechanism for fibrosis in various organs including skin [23]. Recent studies detected EMT in intestinal fibrosis [13, 23]. However, the actual relevance of EMT in intestinal fibrosis in CD patients stills needs to be further confirmed [24].
IL-17A is a pro-inflammatory cytokine. It is mainly produced and secreted by TH17 cells. Abnormal expression of IL-17A is believed to be potential diagnostic markers for initiation and development of a number of acute and chronic diseases. In recent years, studies on IL-17A have gone beyond its pro-inflammatory activities. It has been shown to participate in fibrotic process of multiple organs [25,26,27] including intestinal fibrosis. In TNBS-induced mouse models of acute and chronic colitis, the administration of vaccines against IL-23 was found to significantly reduce collagen deposition. It is speculated that this effect is due to reduction in TH17 cells [28]. The mechanisms for IL-17A’s pro-fibrotic effects may come from its activation of fibroblasts that lead to secretion and deposition of large amount of ECM. More recent reports indicate that IL-17A may induce EMT on epithelial cells to promote fibrosis indirectly [29]. The questions are how does IL-17A change in CD and what association does IL-17A have with EMT? In this study, we will focus on the roles of IL-17A and EMT in intestinal fibrosis.
Materials and Methods
Ethics Statement
The research involved human participants and was conducted according to the principles described in the Declaration of Helsinki. The Medical Ethics Committee of China Medical University approved this study (protocol number: AF-SOP-07-1, 1-01). All patients participating in the study signed the informed consent form for Scientific Use of Biological Materials in accordance with requirements set by the ethics committee. All the samples were anonymous.
Clinical Patients and CD Tissue Samples
All clinical CD samples were biopsy tissues collected at intestinal stricture sites of patients who were diagnosed as CD by colonoscopy at the First Affiliated Hospital of China Medical University from August 2012 to March 2013. In total, there were 14 patients, among whom 10 were female and four were male. Clinical and pathological findings in all 14 cases met the criteria for CD diagnosis. Samples of control group were collected from normal intestinal mucosal tissue adjacent to intestinal polyps of eight patients who were diagnosed by colonoscopy during the same period. Among them four were male and four were female. CT examination was performed in all clinical cases. We chose the cases which showed abnormally thickened loop with minimal enhancement, and no adjacent mesenteric inflammation at the sites of intestinal stenosis in CT which probably suggesting the intestinal fibrosis [30] as CD group. Eight patients in the control group performed the CT scan after colonoscopy, and there were no intestinal stenosis and thickening of the intestine wall in the control group. None of the above patients had received treatment of immunosuppressants within 3 months prior to tissue collection. Clinical features of patients are given in Additional file: Table S1 + 2. Biopsy tissues were immediately frozen in liquid nitrogen after collection and later transferred into − 80 °C freezer for storage. Clinical information and data were collected anonymously.
Cell Culture
Rat normal intestinal crypt epithelial cell line IEC-6 was provided by the Cell Resources Center of the Institute of Basic Medical Sciences (IBMS) of the Chinese Academy of Medical Sciences (CAMS). The cells were cultured in DMEM-H media supplemented with 10% heat-inactivated calf serum and 0.01 mg/ml insulin and incubated at 37 °C and 5% CO2 conditions.
Cell Treatment
Log-phase growing cells were inoculated in 6-well plates with 8 × 105 cells per 2 ml media per well at 37 °C, 5% CO2; once cell confluency reached 30–50%, old media were replaced by new DMEM-H media supplemented with 1% FBS for 24 h; stimulating factor TGF-β1 (Peprotech, USA) was added to 10 ng/ml and IL-17A (Peprotech, USA) was added to 10, 50 and 100 ng/ml; cell morphology was observed after 72 h under an inverted microscope; and cell lysates were harvested.
Establishment of Animal Model
Male BALB/c mice between 6 and 8 weeks old weighing 20 ± 2 g were obtained from Experimental Animal Center of China Medical University. The mice were randomly divided into five groups with 10 mice per group and were determined as specific pathogen-free, housed on a 12:12-h light: dark cycle in sterile micro-isolator cages, and fed ad libitum on standard chow. Enema method [31]: Food (but not water) was withdrawn 24 h prior to enema. Mice were anesthetized with peritoneal injection of 4% (10 ml/kg) of chloral hydrate. Fifty microliters of enema solution was administered via a polyethylene catheter that was advanced through the rectum into the colon until the tip was 1 cm proximal to the anus. Another 100 µl of solution was injected after catheter was advanced 4 cm into the colon. To ensure the retention of the solution within the entire colon and cecum, mice were held by tail in a vertical position for 30 s after the intrarectal administration. In mice that were given TNBS (Sigma-Aldrich, St. Louis, MO, USA), 1 week prior to enema, a 2 cm × 2 cm abdominal skin area was exposed by shaving the hair and TNBS solution was applied to the skin area for sensitization. Treatments in each group (10 mice per group): (1): normal control group: regular food and water, no special treatment; (2) EtOH control group: 150 μl of 45% ethanol was used as enema solution once per week for 6 weeks consecutively; (3) TNBS group: increasing concentrations of TNBS solutions were used for enema [32]: 1st and 2nd weeks, 150 μl of 1.5 mg/0.1 ml TNBS/45% ethanol solution once per week; 3rd and 4th weeks, 150 μl of 2.0 mg/0.1 ml TNBS/45% ethanol solution once per week; 5th and 6th weeks, 150 μl of 2.5 mg/0.1 ml TNBS/45% ethanol solution once per week; (4) anti-IL-17A-neutralizing antibody treatment group: the mice received enema treatment the same way as those did in TNBS model group. On the 22nd, 29th, and 36th days, 2 μg of anti-IL-17A-neutralizing antibody (Purified Rat Anti-Mouse IL-17A Clone, BD Biosciences, USA) was given to each mouse via tail vein injection 2 h prior to TNBS enema; (5) IgG isotype treatment group: the mice received enema treatment the same way as those did in TNBS model group. On the 22nd, 29th, and 36th days, 2 μg of isotype IgG (Purified Rat IgG Isotype Control, BD Biosciences, USA) was given to each mouse via tail vein injection 2 h prior to TNBS enema. In normal and EtOH control groups, mice were injected with same volumes of normal saline solution.
During the course of experiment, mice were closely monitored on a daily basis for food intake, water drinking, and other regular activities. Body weight, characteristics of stool, occult blood, and blood in stool were recorded for DAI scoring [33] (Table S3). Clinical symptoms were used for evaluation on the severity of colitis [34].
Collection and Handling of Specimen
All mice were killed by cervical dislocation at the end of experiment after the 6th week. Under sterile condition, a midline laparotomy was performed and whole colon was cut out. Macroscopic assessment of colonic inflammation and fibrosis was made by examining conditions of colonic wall thickening, congestion, stricture, adhesion to surrounding tissues, and measurement of lesion lengths [35]. After vigorous rinse with PBS, we cut the colonic tissue 2 cm away from the anus as total and then divided this colorectal segment into two equal parts: the first segment which was proximal to the anus was snap-frozen in liquid nitrogen and later stored in − 80 °C freezer for subsequent collagen estimation and Western blot, and the second segment which was far from the anus was fixed in 4% paraformaldehyde for hematoxylin and eosin (HE) and Masson trichrome stain (MTS).
Histological Assessment of Colitis and Colonic Fibrosis
After fixation with paraformaldehyde, colonic tissue specimen was embedded in paraffin and was cut to 4 μm sections. Standard HE and MTS (Masson trichrome stain) were performed [36], and the degree of colitis and fibrosis was scored [37].
Collagen Estimation by Hydroxyproline Quantification
Sixty milligrams of colonic tissue was hydrolyzed in 2.5 N NaOH for 40 min at 120 °C, 0.1 kPa. After neutralization by HCl, the hydrolysate was diluted by distilled water. The hydroxyproline content of the hydrolyzation products was assessed colorimetrically at 550 nm with p-dimethylaminobenzaldehyde. The results were expressed as μg per mg of colonic tissue [37].
Real-Time PCR
We examined the mRNA levels expression of EMT-related genes in clinical biopsy specimen and post-treatment cells. In brief, total RNA from tissue and cells was extracted using TRIzol kit (Invitrogen, USA) following the manufacturer’s manual and later reversed-transcribed into cDNAs by using a reverse transcription kit (Takara, Japan). Those cDNAs were then used as templates for amplification with real-time PCR assay kit (Takara, Japan) and being analyzed in 7500 fast real-time PCR system (ABI). Primers for the real-time PCR amplification are as follows:
Human IL-17A: Forward 5′-TCAACCCGATTGTCCACCAT-3′, Reverse 5′-GAGTTTAGTCCGAAATGAGGCTG -3′; E-cadherin: Forward 5′-GAGTGCCAACTGGACCATTCAGTA-3′, Reverse 5′-AGTCACCCACCTCTAAGGCCATC-3′; vimentin: Forward 5′-AGGAACAGCATGTCCAAATCG-3′, Reverse 5′- AAGGGCATCCACTTCACAGG-3′; snail: Forward 5′-CGAAAGGCCTTCAACTGCAAAT-3′, Reverse 5′- ACTGGTACTTCTTGAACTCTG -3′; α-SMA: Forward 5-ATAACATCAAGCCCAAATCTGC-3′, Reverse 5′-TTCCTTTTTTCTTTCCCAACA-3′; β-actin: Forward 5′-TGGCACCCAGCACAATGAA-3′, Reverse 5′-CTAAGTCATAGTCCGCCTAGAAGCA -3′.
Western Blot
Proteins from colonic tissue and IEC-6 cells were extracted according to standard protocols. Protein concentrations were quantified using BCA (bicinchoninic acid) method. Total proteins were separated through SDS-PAGE gel system and were later transferred onto PVDF membrane, which was blocked and then incubated with primary antibodies for E-cadherin (1:2000, BD BioSciences, CA, USA), vimentin (1:1000, Cell Signaling Technology, Danvers, MA, USA), snail (1:1000, Cell Signaling Technology, Danvers, MA, USA), FSP1 (1:400, Abcam, HK), α-SMA (1:1000, Boster ImmunoLeader, China), IL-17A (1:400, Bioss, China), β-actin (1:5000, Cell Signaling Technology, Danvers, MA, USA), followed by incubation with corresponding secondary antibodies. Proteins were visualized by enhanced chemiluminescence (ECL) kit (PerkinElmer, USA), and images were captured by a Bio-Rad imaging system.
Statistical Analysis
Data were analyzed using SPSS 16.0 software. Results from each treatment group were presented as mean ± SD, and comparisons among groups were first made by homogeneity of variances test. If the test was passed, one-way analysis of variance test (ANOVA) was applied to check the overall differences between means, and if there was significant difference, a multiple comparison post hoc test (Student–Newman–Keuls) was performed to evaluate differences between the experimental groups. If the homogeneity test was not passed, a nonparametric test was then performed. For all analyses, p < 0.05 was considered to be statistically significant.
Results
Expression of IL-17A and EMT-Related Genes in Colonic Mucosal Biopsy Tissues of CD Patients and Control Individuals
Using real-time PCR, we first examined at transcription levels of the gene expression of IL-17A and EMT-related indicators (E-cadherin, vimentin, snail, α-SMA) in colonic mucosal biopsy tissues of CD patients and control individuals. As shown in Fig. 1, mRNA levels of IL-17A in colonic mucosal tissue of CD patients were significantly higher than those in control groups (p < 0.01); Among EMT-related genes, compared with those in control groups, mRNA levels of E-cadherin in colonic mucosal tissues from CD patients were significantly lower (p < 0.01), while mRNA levels of vimentin and α-SMA were remarkably increased (p < 0.05, p < 0.01, respectively). There were no significant differences in expression of snail between CD and control groups (p > 0.05). Later, we used Western blot to examine protein expression of IL-17A and EMT-related genes in colonic mucosal biopsy tissues of CD patients and control individuals. Consistent with findings at mRNA level, Western results show (Fig. 2) that protein expression of IL-17A in mucosal tissue of CD patients was significantly higher than those in control individuals (p < 0.01); among EMT-related genes, E-cadherin protein expression was significantly lower in CD colonic mucosal tissue than those in control tissues (p < 0.01), while protein expression of vimentin, snail, and α-SMA was significantly higher than those in control groups (p < 0.05, p < 0.05, p < 0.01, respectively).

Transcription levels of the gene expression of IL-17A and EMT-related genes in colonic mucosal biopsy tissues of control individuals and CD patients. *p < 0.05, **p < 0.01 versus control individuals

Protein expression of IL-17A and EMT-related genes in colonic mucosal biopsy tissues of CD patients and control individuals. a, c The E-cadherin, vimentin, snail, IL-17A, and α-SMA protein bands of Western blot detection. b, d Gray value histogram of Western blot detection. The fold change of target protein was normalized to control. Values mean ± SD, *p < 0.05, **p < 0.01 versus control individuals
IL-17A-Induced EMT Process In Vitro
Our study on clinically collected tissues found occurrence of EMT in CD-affected colonic mucosa with concomitant high expression of IL-17A in those same tissues which may suggest that IL-17A could promote intestinal fibrosis through its induction of EMT process in mucosal cells. To verify the potential inductive effect of IL-17A on EMT, we cultured IEC-6 cells for 72 h under increasing concentrations of IL-17A or with 10 ng/ml of TGF-β1. Results show that the control IEC-6 cells were well connected with intercellular adhesion molecules and tight junction and exhibited homogenous cell density and morphology; after 72-h treatment with 10 ng/ml IL-17A, a small portion of intercellular tight junction disappeared. Under 50 or 100 ng/ml of IL-17A treatment for 72 h more IEC-6 cells lost tight junction and their morphology transformed into more irregular and spindle shape. Various degrees of tight junction loss were also seen in cells under 10 ng/ml of TGF-β1 stimulation, and these cells started to adopt mesenchymal cell morphology (Fig. 3a). At protein levels, Western blot was used to examine the expression of classical EMT indicators vimentin, snail, and E-cadherin (Fig. 3b). Meanwhile, we also examined the expression of two fibroblast marker proteins that were used to identify sources of EMT: FSP1 and α-SMA (Fig. 3b), which were also important indicators for fibrosis. Results show (Fig. 3c) that IL-17A was able to induce the expression of vimentin, snail, FSP1, and α-SMA while inhibit the expression of E-cadherin. In particular, 100 ng/ml of IL-17A demonstrated the strongest induction of EMT on IEC-6 cells.

In vitro effect of IL-17A on EMT. a Light microscopy analysis shows the morphologic changes of IEC-6 cells treated with IL-17A or with TGF-β1 (magnification, × 100). (I) the control group, (II) the model group (treated with 10 ng/ml IL-17A), (III) the model group (treated with 50 ng/ml IL-17A), (IV) the model group (treated with 100 ng/ml IL-17A), (V) the positive control group (treated with 10 ng/ml TGF-β1). b Effects of IL-17A on protein expression of classical EMT indicators and fibroblast markers. c Gray value histogram of Western blot detection. Values mean ± SD. *p < 0.05, **p < 0.01 versus control group
Blockage of IL-17A In Vivo Attenuated TNBS-Induced Experimental Colitis and Colonic Fibrosis
Clinical Symptoms and Survival Rates of Experimental Mice
During experiments, mice in normal and EtOH control groups showed good general appearance, glossy hair coat, being alert and actively foraging, continuous weight gaining; in mice of TNBS and isotype IgG treatment group, various degrees of symptoms started on day 1 and intensified on day 2 after TNBS enema: inactivity or lethargy, hunched back, diarrhea, mucous in stool, occult blood in stool, reduced food intake with increased water drinking. Most mice lost weight 2–4 days after each of the first three enemas followed by gradually regaining some body weight afterward. Some mice may even gain more weight than they had before enema. With increase in number of enemas, by the 4th week, the symptoms became stabilized with improved stool characteristics and increased body weight in most animals. Mice in anti-IL-17A treatment group showed similar symptoms in the first 3 weeks but had significant improvements after 4th week. DAI scores for mice in each group on day 2 after each weekly enema are shown in Fig. 4. Scores of TNBS and isotype IgG groups are significantly higher than those of normal and EtOH control groups (p < 0.01). Scores of anti-IL-17A treatment group are significantly higher than those in the control groups during the first 3 weeks, but from the 4th week (i.e., time when anti-IL-17A treatment started) the scores become significantly lower than those in TNBS and isotype IgG groups (p < 0.01). No statistical differences were found between normal and EtOH control groups or between TNBS and isotype IgG groups (p > 0.05).

DAI scores for mice in each group on day 2 after each weekly enema. Scores of TNBS and isotype IgG groups are significantly higher than those of control and EtOH control groups (p < 0.01). Scores of anti-IL-17A treatment group are significantly higher than those in the control groups during the first 3 weeks, but from the 4th week (i.e., time when anti-IL-17A treatment started) the scores become significantly lower than those in TNBS and isotype IgG groups (p < 0.01). No statistical differences were found between control and EtOH control groups or between TNBS and isotype IgG groups (p > 0.05)
TNBS, anti-IL-17A, and isotype IgG groups all had mouse deaths. The mortality rate is 20, 10, and 20%, respectively. Relatively high mortality rates occurred during the first 3 weeks, and mortality rates went down by 4th week. Autopsy showed colons in most mice had severe edema, hemorrhage, necrosis, and in rare cases of perforation. Mice in normal and EtOH control groups had no obvious changes in regular activities. Most of these mice gained body weight continuously. There were no deaths in these two control groups.
Macroscopic Changes in Colonic Tissues of Mice from Each Group
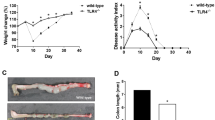
Colonic mucosa in normal control mice was smooth and free from edema or congestion; slight colonic mucosa congestion was observed in only one mouse in EtOH control group; induced chronic colitis model in TNBS, anti-IL-17A, and isotype IgG groups shares features of CD: Lesions occurred in both colon and rectum, the affected colonic mucosa had edema and congestion, thickening of colonic wall and local colonic wall became stiff and twisted. In some mice, adhesion of intestinal wall to surrounding tissue was observed (Fig. 5a). Colon length of mouse in each group is shown in Fig. 5b: Colon length in TNBS, anti-IL-17A, and isotype IgG groups was all significantly shorter than normal and EtOH control groups (p < 0.01); however, length in anti-IL-17A group was significantly longer than those in TNBS and isotype IgG groups (p < 0.01); macroscopic assessment of mouse colon in each group is shown in Fig. 5c: Comparing with normal and EtOH control groups, scores in TNBS, anti-IL-17A, and isotype IgG groups were all significantly increased (p < 0.01); however, scores in anti-IL-17A group were significantly lower than those in TNBS and isotype IgG groups (p < 0.01).

Effect of blocking IL-17A on macroscopic changes in colonic tissues of mice: a macroscopic appearance of colons from each group. Colonic mucosa in normal control and EtOH control mice was smooth and free from edema or congestion; induced chronic colitis model in TNBS, anti-IL-17A, and isotype IgG groups shares features of CD: the affected colonic mucosa had edema and congestion, thickening of colonic wall and local colonic wall became stiff and twisted. In some mice, adhesion of intestinal wall to surrounding tissue was observed. b Colon length of each group. c Macroscopic score of each group. Values mean ± SD. *p < 0.01 versus control and EtOH control group, *p < 0.01 versus *#
Microscopic Findings in Colonic Tissues of Mice of Each Group
HE staining of mouse colonic tissue (Fig. 6a): In normal control group, the integrity of intestinal mucosa was intact, the intestinal glands composed of single column epithelium, lamina propria, and muscularis mucosae were well aligned; in EtOH control group, alignment of intestinal glands was intact although infiltration of inflammatory cells was seen in some parts of mucosa and submucosa; in TNBS and isotype IgG groups, there were loss of colonic crypts and goblet cells, fragmented epithelium, mucosal erosion and ulceration; infiltration of inflammatory cells in all layers of colon, particularly in the mucosa and submucosa, the infiltrated inflammatory cells were mainly lymphocytes and monocytes which was typical histological features of chronic colitis; in anti-IL-17A treatment group, although lesions were seen in colonic tissues, the size, depth, and extent of inflammatory cell infiltration were much less than those in TNBS and isotype IgG groups. Compared with normal and EtOH control groups, scores of pathological evaluation in TNBS, anti-IL-17A treatment, and isotype IgG groups were all significantly higher (p < 0.01); however, score in anti-IL-17A was significantly lower than those in TNBS and isotype IgG groups (p < 0.01). Score in EtOH control group was higher than that in normal control group, but there were no significant differences.

Effect of blocking IL-17A on microscopic changes in colonic tissues of mice of each group (a, control group; b, EtOH control group; c, TNBS group; d, TNBS + IL-17Ab group; e, TNBS + IgG group): A Light-field microscopic histological analysis in experimental fibrosis stained with hematoxylin and eosin (magnification, × 40). Microscopic score of each group, values mean ± SD. *p < 0.01 versus control and EtOH control group, *p < 0.01 versus *# B Light-field microscopic analysis of Masson’s trichrome-stained colon sections of each group (magnification, × 40). Fibrosis score of each group, values mean ± SD. *p < 0.01 versus control and EtOH control group, *p < 0.01 versus *#
Masson staining in mouse colonic tissue (Fig. 6b): In TNBS and isotype IgG groups, there was large amount of blue-stained collagen deposition in mucosa and serosa layers with occasional fibrous septa. In anti-IL-17A group, sporadic deposition of collagen fibers was seen in the above areas. No collagen deposition was seen in normal or EtOH control groups. Compared with normal and EtOH groups, scores for mouse colonic fibrosis in TNBS, anti-IL-17A, and isotype IgG groups were significantly higher (p < 0.01); however, degree of fibrosis in anti-IL-17A was significantly less than those in TNBS and isotype IgG groups (p < 0.01).
Hydroxyproline Contents in Mouse Colonic Tissue of Each Group
Hydroxyproline (Hyp) accounts for around 13.4% of amino-acids composition in collagen. There is only tiny amount of Hyp in elastin and none in other proteins. Majority of collagen is distributed in skin, cartilage, and vasculatures. Therefore, content of Hyp is a reliable marker for collagen deposition in connective tissue disorders. As shown in Fig. 7, compared with normal and EtOH control groups, Hyp was significantly increased in TNBS, anti-IL-17A, and isotype IgG groups (p < 0.05), while Hyp in anti-IL-17A was significantly lower than those in TNBS and isotype IgG groups (p < 0.01).

Effect of blocking IL-17A on hydroxyproline contents in mouse colonic tissues of each group. Content of Hyp is a reliable marker for collagen deposition in connective tissue disorders. *p < 0.05 versus control and EtOH control group, *p < 0.01 versus *#
Expression of EMT- and Fibrosis-Related Proteins in Mouse Colonic Tissue of Each Group
Compared with normal and EtOH groups, E-cadherin protein expressions in TNBS, isotype IgG, and anti-IL-17A groups were decreased (p < 0.05), while vimentin and snail protein expressions were increased (p < 0.05); however, protein expression of E-cadherin in anti-IL-17A was higher than those in TNBS and isotype IgG groups (p < 0.05), while protein expressions of vimentin and snail were lower than those in TNBS and isotype IgG groups (p < 0.05). Compared with normal and EtOH control groups, protein expressions of fibrosis marker α-SMA in TNBS, isotype IgG, and anti-IL-17A were increased (p < 0.05); however, α-SMA expression in anti-IL-17A was lower than that in TNBS and isotype IgG (p < 0.05) (Fig. 8).

Effect of blocking IL-17A on expression of EMT- and fibrosis-related proteins in mouse colonic tissue of each group (I, control group; II, EtOH control group; III, TNBS group; IV, TNBS + IL-17Ab group; V, TNBS + IgG group). a The E-cadherin, vimentin, snail, and α-SMA protein bands of Western blot detection. b Gray value histogram of Western blot detection. Values mean ± SD. *p < 0.05 versus control and EtOH control group, *p < 0.05 versus *#
Discussion
Fibrosis is a common complication in IBD and is considered as an important cause of intestinal stricture in CD patients. The lack of knowledge in the mechanisms of intestinal fibrosis is one of the major obstacles in the development of effective anti-fibrotic drugs. Therefore, it has now been suggested that multiple mechanisms could be responsible for reversing fibrosis on a molecular level in IBD [38].
In intestinal fibrosis, the main progenitor cells of activated myofibroblasts are the fibroblasts and the epithelial cells, and the latter is through the process of EMT [16, 39, 40]. Our study also found alterations in expression of classical indicators of EMT in intestinal mucosal tissue of CD patients (E-cadherin, vimentin, and α-SMA) except snail. Snail is a specific transcriptional factor which can recognize the E-box sequence in its target gene promoter, thus inhibiting the target gene expression. The promoter region of E-cadherin gene also contains E-box sequences; therefore, snail may downregulate E-cadherin expression [41]. Upregulation of snail expression during EMT process may help clearly identify the occurrence of EMT. In our study, we found that transcriptional levels of snail in CD group had an increasing trend over control group, though it lacked statistical significance, while at protein expression level it was significantly higher in CD group than that in control group. The discrepancy between the transcription and protein levels may be due to the following reasons: (1) number of cases was not enough, no statistical significance due to small sample size; (2) posttranscriptional processing, transcriptional products undergoes degradation, translation, and posttranslational modification; therefore, results at transcription and protein levels may not be exactly consistent; (3) space and time interval during eukaryotic gene transcription and translation, due to detection at different time points, mRNA might have been degraded when protein expression was at its peak or protein expression was still increasing when mRNA had reached its peak. In summary, our study confirmed the involvement of EMT in intestinal fibrosis in CD.
IL-17A is linked to the development of intestinal fibrosis [42]. Our study also confirmed the involvement of IL-17A in development of intestinal fibrosis. Recent studies found that IL-17A may promote the development of fibrosis by inducing EMT in alveolar epithelial cells and obstructive bronchitis [43, 44]. Is EMT one of the pathways through which IL-17A promotes intestinal fibrosis in CD? Our results of cells showed that IL-17A repressed expression of E-cadherin but promoted expression of vimentin and FSP1. Reduction of E-cadherin expression will lead to loss of intercellular tight junction and changes in cellular polarity. Highly expressed vimentin suggests that epithelial cells are transformed into mesenchymal cells and cell morphology become spindly, and vimentin also may downregulate E-cadherin since it can block transport of E-cadherin onto cell surface [45]. FSP1-positive fibroblasts are usually derived from epithelial through EMT; therefore, it is often used as a marker for EMT-derived fibroblasts. Moreover, our study found increased expression of α-SMA in IL-17A-treated cells. α-SMA has been recognized as a key marker for differentiation and activation of myofibroblasts. Aggregation and activation of large number of myofibroblasts can produce and secret large amount of ECM [39, 46]. The above results indicate that IL-17A may transform epithelial cells into fibroblasts and activated myofibroblasts via EMT. This simplified model can be used to a certain extent for in vitro study of intestinal fibrosis.
To further test the hypothesis that IL-17A promotes intestinal fibrosis through inducing EMT, we used the mouse model of TNBS enema-induced experimental colitis and colonic fibrosis for our in vivo study. In the mouse model of chronic colitis, elevation of IL-17A began from the 3rd to the 4th week and continued till the 42nd day to reach its peak [32, 47, 48]. For the above reasons, we established the mouse model of intestinal fibrosis by repeatedly giving mice enema for 6 weeks with increased concentration of TNBS, and starting from the 4th week on mice were injected with anti-IL-17A antibody to neutralize IL-17A in their bodies. Following anti-IL-17A treatment, the changes in the results (DAI scores, colon length, macroscopic scores, microscopic scores, fibrosis scores, and the content of Hyp) indicate that neutralization of IL-17A might have inhibited TNBS-induced inflammatory reaction and fibrosis. The expression of protein levels of the classical markers of EMT and fibrosis indicates that anti-IL-17A antibody may inhibit intestinal fibrosis development through repressing EMT at the molecular level, thus further confirming that IL-17A may promote development of intestinal fibrosis through the pathway of EMT.
In conclusion, our results indicate that EMT process is one of the pathways of IL-17A uses to promote intestinal fibrosis both in clinical CD patients and in TNBS-induced chronic colitis. It could represent a valid new target for the development of anti-fibrotic drugs, possibly by blocking the IL-17A activity. But recently non-exciting results reported that blockade of either IL-17A (secukinumab) or its receptor (brodalumab) was ineffective and secukinumab may even worsen disease in an RCT of moderate-to-severe CD. Due to the reason that IL-17A has been claimed to show both a protective and exacerbating effect in preclinical murine models, they point out that blocking IL-17A/IL-17R may interfere with a protective function of IL-17A in the intestine [49]. While other observation that IL-17A expression is higher in long-standing CD mucosa compared with early mucosal lesions [50], and intestinal fibrosis is a late-stage process in CD, so even if blocking IL-17A may worsen inflammatory disease in moderate-to-severe CD, we cannot deny the role of anti-IL-17A antibody in counteracting the fibrogenic process in CD; therefore, we need further studies to clarify the therapeutic effect of blocking IL-17A in intestinal fibrosis in CD.
References
Latella G, Di Gregorio J, Flati V, Rieder F, Lawrance IC. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol. 2015;50:53–65.
Jeuring S, Van den Heuvel T, Zeegers M, et al. Disease behavior in Crohn’s disease patients diagnosed in the biological era—a Dutch population-based IBD-SL cohort study. Gastroenterology. 2015;148:2.
Cosnes J, Bourrier A, Nion-Larmurier I, Sokol H, Beaugerie L, et al. Factors affecting outcomes in Crohn’s disease over 15 years. Gut. 2012;61:1140–1145.
Pittet V, Rogler G, Michetti P, Fournier N, Vader JP, et al. Penetrating or stricturing diseases are the major determinants of time to first and repeat resection surgery in Crohn’s disease. Digestion. 2013;87:212–221.
Latella G, Papi C. Crucial steps in the natural history of inflammatory bowel disease. World J Gastroenterol. 2012;18:3790–3799.
Bernstein CN, Loftus EV Jr, Ng SC, Lakatos PL, Moum B, et al. Hospitalisations and surgery in Crohn’s disease. Gut. 2012;61:622–629.
Latella G, Sferra R, Vetuschi A, Zanninelli G, D’Angelo A, et al. Prevention of colonic fibrosis by Boswellia and Scutellaria extracts in rats with colitis induced by 2,4,5-trinitrobenzene sulphonic acid. Eur J Clin Invest. 2008;38:410–420.
Speca S, Giusti I, Rieder F, Latella G. Cellular and molecular mechanisms of intestinal fibrosis. World J Gastroenterol. 2012;18:3635–3661.
Rieder F, Fiocchi C. Intestinal fibrosis in inflammatory bowel disease—current knowledge and future perspectives. J Crohns Colitis. 2008;2:279–290.
Rieder F, Fiocchi C, Rogler G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology. 2017;152:e346.
Lawrance IC, Rogler G, Bamias G, Breynaert C, Florholmen J, et al. Cellular and molecular mediators of intestinal fibrosis. J Crohns Colitis. 2017;11:1491–1503.
Rieder F. The gut microbiome in intestinal fibrosis: Environmental protector or provocateur? Sci Transl Med. 2013;5:190ps110.
Scharl M, Huber N, Lang S, Furst A, Jehle E, et al. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn’s disease associated intestinal fibrosis. Clin Transl Med. 2015;4:1.
Scharl M, Frei S, Pesch T, Kellermeier S, Arikkat J, et al. Interleukin-13 and transforming growth factor beta synergise in the pathogenesis of human intestinal fistulae. Gut. 2013;62:63–72.
Flier SN, Tanjore H, Kokkotou EG, Sugimoto H, Zeisberg M, et al. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem. 2010;285:20202–20212.
Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. 2011;179:2660–2673.
Rieder F, Brenmoehl J, Leeb S, Scholmerich J, Rogler G. Wound healing and fibrosis in intestinal disease. Gut. 2007;56:130–139.
Uehara H, Nakagawa T, Katsuno T, Sato T, Isono A, et al. Emergence of fibrocytes showing morphological changes in the inflamed colonic mucosa. Dig Dis Sci. 2010;55:253–260. https://doi.org/10.1007/s10620-009-0730-7.
Brittan M, Chance V, Elia G, Poulsom R, Alison MR, et al. A regenerative role for bone marrow following experimental colitis: contribution to neovasculogenesis and myofibroblasts. Gastroenterology. 2005;128:1984–1995.
Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt. Cell. 2016;166:21–45.
Grigore AD, Jolly MK, Jia D, Farach-Carson MC, Levine H. Tumor budding: the name is EMT. Partial EMT. J Clin Med. 2016;5:51.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196.
Yang J, Zhou CZ, Zhu R, Fan H, Liu XX, et al. miR-200b-containing microvesicles attenuate experimental colitis associated intestinal fibrosis by inhibiting epithelial-mesenchymal transition. J Gastroenterol Hepatol. 2017;32:1966–1974.
Scharl M, Bruckner RS, Rogler G. The two sides of the coin: similarities and differences in the pathomechanisms of fistulas and stricture formations in irritable bowel disease. United Eur Gastroenterol J. 2016;4:506–514.
Okamoto Y, Hasegawa M, Matsushita T, Hamaguchi Y, Huu DL, et al. Potential roles of interleukin-17A in the development of skin fibrosis in mice. Arthritis Rheum. 2012;64:3726–3735.
Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. 2010;207:535–552.
Tan Z, Qian X, Jiang R, Liu Q, Wang Y, et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol. 2013;191:1835–1844.
Guan Q, Ma Y, Hillman CL, Qing G, Ma AG, et al. Targeting IL-12/IL-23 by employing a p40 peptide-based vaccine ameliorates TNBS-induced acute and chronic murine colitis. Mol Med. 2011;17:646–656.
Latella G, Caprilli R, Travis S. In favour of early surgery in Crohn’s disease: a hypothesis to be tested. J Crohns Colitis. 2011;5:1–4.
Quencer KB, Nimkin K, Mino-Kenudson M, Gee MS. Detecting active inflammation and fibrosis in pediatric Crohn’s disease: prospective evaluation of MR-E and CT-E. Abdominal Imaging. 2013;38:705–713.
Scheiffele F, Fuss IJ. Induction of TNBS colitis in mice. Curr Protoc Immunol. 2002;49:19.
Fichtner-Feigl S, Fuss IJ, Young CA, Watanabe T, Geissler EK, et al. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol. 2007;178:5859–5870.
Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352.
Dieleman LA, Pena AS, Meuwissen SG, van Rees EP. Role of animal models for the pathogenesis and treatment of inflammatory bowel disease. Scand J Gastroenterol Suppl. 1997;223:99–104.
Videla S, Vilaseca J, Medina C, Mourelle M, Guarner F, et al. Selective inhibition of phosphodiesterase-4 ameliorates chronic colitis and prevents intestinal fibrosis. J Pharmacol Exp Ther. 2006;316:940–945.
Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546.
Lawrance IC, Wu F, Leite AZ, Willis J, West GA, et al. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology. 2003;125:1750–1761.
Bettenworth D, Rieder F. Reversibility of stricturing Crohn’s disease-fact or fiction? Inflamm Bowel Dis. 2016;22:241–247.
Fiocchi C, Lund PK. Themes in fibrosis and gastrointestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2011;300:G677–G683.
Rieder F, Fiocchi C. Mechanisms of tissue remodeling in inflammatory bowel disease. Dig Dis. 2013;31:186–193. https://doi.org/10.1159/000353364.
Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428.
Biancheri P, Pender SL, Ammoscato F, Giuffrida P, Sampietro G, et al. The role of interleukin 17 in Crohn’s disease-associated intestinal fibrosis. Fibrogenesis Tissue Repair. 2013;6:13.
Mi S, Li Z, Yang HZ, Liu H, Wang JP, et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol. 2011;187:3003–3014.
Vittal R, Fan L, Greenspan DS, Mickler EA, Gopalakrishnan B, et al. IL-17 induces type V collagen overexpression and EMT via TGF-beta-dependent pathways in obliterative bronchiolitis. Am J Physiol Lung Cell Mol Physiol. 2013;304:L401–L414.
Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851.
Mifflin RC, Pinchuk IV, Saada JI, Powell DW. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol. 2011;300:G684–G696.
Zhu MY, Lu YM, Ou YX, Zhang HZ, Chen WX. Dynamic progress of 2,4,6-trinitrobenzene sulfonic acid induced chronic colitis and fibrosis in rat model. J Dig Dis. 2012;13:421–429.
Fichtner-Feigl S, Strober W, Geissler EK, Schlitt HJ. Cytokines mediating the induction of chronic colitis and colitis-associated fibrosis. Mucosal Immunol. 2008;1:S24–S27.
Verstockt B, Ferrante M, Vermeire S, Van Assche G. New treatment options for inflammatory bowel diseases. J Gastroenterol. 2018;53:585–590.
Zorzi F, Monteleone I, Sarra M, Calabrese E, Marafini I, et al. Distinct profiles of effector cytokines mark the different phases of Crohn’s disease. Gastroenterology. 2013;144:S820–S820.
Acknowledgments
This work was supported by Liaoning Province Science and Technology Project (2013225049). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
MJ-S was involved in conception and design, interpretation of data, and manuscript revision. HJ-Z, YN-Z, and HZ performed the experiments. LG and YL collected the biopsy samples. HJ-Z YN-Z analyzed the data. MJ-S contributed reagents/materials/analysis tools. HJ-Z and YN-Z wrote the paper. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors do not have a commercial or other association that might pose a conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, HJ., Zhang, YN., Zhou, H. et al. IL-17A Promotes Initiation and Development of Intestinal Fibrosis Through EMT. Dig Dis Sci 63, 2898–2909 (2018). https://doi.org/10.1007/s10620-018-5234-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-018-5234-x