
Abstract
Fibrosis is an important pathological change in inflammatory bowel disease (IBD), but the mechanism has yet to be elucidated. WNT2B high‑expressed fibroblasts are enriched in IBD intestinal tissues, although the precise function of this group of fibroblasts remains unclear. This study investigated whether WNT2B high‑expressed fibroblasts aggravated intestinal tissue damage and fibrosis. Our study provides evidence that WNT2B high‑expressed fibroblasts and NK cells were enriched in colitis tissue of patients with IBD. WNT2B high‑expressed fibroblasts secreted wnt2b, which bound to FZD4 on NK cells and activated the NF-κB and STAT3 pathways to enhance IL-33 expression. TCF4, a downstream component of the WNT/β-catenin pathway, bound to p65 and promoted binding to IL-33 promoter. Furthermore, Salinomycin, an inhibitor of the WNT/β-catenin pathway, inhibited IL-33 secretion in colitis, thereby reducing intestinal inflammation.Knocking down WNT2B reduces NK cell infiltration and IL-33 secretion in colitis, and reduce intestinal inflammation and fibrosis. In conclusion, WNT2B high‑expressed fibroblasts activate NK cells by secreting wnt2b, which activates the WNT/β-catenin and NF-κB pathways to promote IL-33 expression and secretion, potentially culminating in the induction of colonic fibrosis in IBD.
Key messages
-
WNT2B high-expressed fibroblasts and NK cells are enriched in colitis tissue, promoting NK cells secreting IL-33.
-
Wnt2b activates NF-κB and STAT3 pathways promotes IL-33 expression by activating p65 and not STAT3. syndrome
-
TCF4 binds to p65 and upregulates the NF- κB pathway.
-
Salinomycin reduces NK cell infiltration and IL-33 secretion in colitis.
-
Knocking down WNT2B mitigates inflammation and fibrosis in chronic colitis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel disease (IBD) is a chronic nonspecific inflammatory disease involving the whole digestive tract. There are two main subtypes of IBD, ulcerative colitis and Crohn’s disease, which share clinical manifestations including fever, diarrhea, abdominal pain, and bloody stools[1]. The pathogenesis of IBD involves heredity, environment, microorganisms, and immunity[2], but at present, it has not been fully elucidated. Intestinal fibrosis is a major pathological feature of inflammatory bowel disease, characterized by excessive extracellular matrix (ECM) protein deposition[3], which is a consequence of chronic inflammation[4], but the precise mechanism remains unclear.
Fibroblasts are normal constituents of the gastrointestinal (GI) tract, mainly participating in the synthesis of ECM and maintaining the integrity of the tissue structure. A variety of fibroblast phenotypes have been recognized. Intestinal tissue predominantly contains fibroblasts, myofibroblasts, and perivascular fibroblasts/pericytes[5]. Myofibroblasts were reported to play a major role in the process of intestinal fibrosis in IBD[6], while the involvement of other clusters remains controversial[7]. It was recently reported that some clusters of fibroblasts termed “inflammatory fibroblasts” could secrete cytokines (IL-6/IL-11)[8] and chemokines (CXCL9/CXCL10/CCL2)[9], thereby promoting angiogenesis and aggravating chronic inflammation[10]. Furthermore, our previous study found another specific subtype of fibroblasts with high expression of WNT2B in IBD[11], which might participate in the inflammatory response; however, the precise function of this group of fibroblasts is unclear.
The involvement of immune cells in the intestinal fibrosis of IBD is important and complex[12]. In intestinal tissue, NK cells are predominantly distributed in the mucous layer, submucosa and Lamina propria[13], directly or indirectly contacting intestinal microorganisms and pathogenic microorganisms and participating in innate and adaptive immune responses[14]. In IBD, NK cells cause an imbalance of the TH1/TH2 response and aggravate the inflammatory response[15], but the precise mechanism of how NK cells are activated and involved in fibrosis are not fully understood. Although it was reported that tumor-associated fibroblasts interfered with the cytotoxicity and cytokine production of NK cells[16], the connection between fibroblasts and NK cells in IBD has yet to be elucidated. The current study aimed to explore the mechanism of interaction between WNT2B high‑expressed fibroblasts and NK cells, which might elucidate a novel mechanism of intestinal fibrosis in inflammatory bowel disease.
Materials and methods
Reagents
The antibodies anti-WNT2B (A19554, 1:1000), anti-GSK3β (A11731, 1:1000), and anti-Phospho-GSK3β-S9 (AP1088, 1:1000) were obtained from Abclonal.Anti-Frizzled4 (ab277797, 2 µg/mL) and Anti-NK1.1 (ab289542, 1:50) were obtained from Abcam.Anti-GAPDH (60,004–1-Ig, 1:20,000), anti-GFP (50,430–2-AP, 1:2000), anti-IL-33 (12,372–1-AP, 1:500), anti-β-catenin (51,067–2-AP, 1:5000), anti-TCF4 (22,337–1-AP, 1:1000), anti-STAT3 (10,253–2-AP, 1:5000), and anti-NF-κB p65 (10,745–1-AP, 1:1000) were from Proteintech. Stat Antibody Sampler Kit II (93130 T, 1:1000), Phospho-Stat Antibody Sampler Kit (9914 T, 1:1000), MAPK Family Antibody Sampler Kit (9926 T, 1:1000), Phospho-MAPK Family Antibody Sampler Kit (9910 T, 1:1000), anti-Akt1 (2938 T, 1:1000), NF-κB Pathway Antibody Sampler Kit (9936 T, 1:1000), and anti-Phospho-Akt (Ser473) (4060 T,1:2000) were from Cell Signaling Technology (CST). Salinomycin (53,003–10-4) was obtained from Selleck.
Plasmid construction
The constructs were from Shanghai Genechem Co.,Ltd.The ΔNLS-TCF4-MYC plasmid mutated both NLS1 and NLS-2 sequences[17].All constructs were verified by DNA sequencing or Western blotting.
Cell lines
Human Intestinal Fibroblast Cells (HIF) were purchased from ScienCell Research Laboratories (CA, USA) and were cultured in Fibroblast Medium (2301, ScienCell) containing 2% fetal bovine serum (0010, ScienCell) and 1% fibroblast growth supplements (2352, ScienCell). NK-92 cells were obtained from American Type Culture Collection (ATCC) (NY, USA) and were maintained in MyeloCult™ H5100 (05150, StemCell Technologies) containing 12.5% horse serum (26,050,088, Gibco) and IL-2 (1:500, 130–097-744, Miltenyi). All cells were maintained at 37 ℃ in a humidified chamber of 5% CO2.
Human colonic tissue
Human colonic tissues were obtained from the normal and inflammatory colonic biopsies of 51 patients with IBD provided by the Department of Gastroenterology and Pathology of Guangzhou Women and Children Medical Center and Guangzhou First People’s Hospital. The ethical aspect of the study was approved by the local ethics committee.
Mice
C57BL/6 J mice were purchased from Southern Medical University (Guangzhou, China). B6.C-Tg(S100a4-cre)1Egn/JhrsJ mice were obtained from The Jackson Laboratory (stock number 030644). Mice were housed at 23 ± 2 °C, a humidity of 55 ± 5%, and a 12-h dark/light cycle. Mice were randomly assigned to different cages for 2 weeks before experiments. All animals were housed and maintained under specific pathogen-free conditions in the animal facility at Nanfang Hospital (Guangzhou, China). All experiments were performed according to the guidelines approved by the Institutional Animal Experiment Committee of Southern Medical University or Nanfang Hospital.
Cell co-culture
HIF transfected with WNT2B plasmids or empty plasmids were co-cultured with NK92 cells in an inserted cell petri dish (14,112, LABSELECT). After 48 h, the culture media were collected to detect the concentration of wnt2b and IL-33. Fibroblasts and NK92 cells were also collected to detect the expression level of corresponding proteins and mRNAs.
CCK-8 assay
A total of 1000–2000 cells in 100ul were added to each well of a 96-well plate. Multiple experimental groups were established, with 3 replicates per group. Cells were incubated at 37℃ in a 5% CO2 cell culture incubator for 12 h. Following the completion of different treatments in each group, 10ul of CCK-8 solution was directly added to each well. The plate containing CCK-8 was incubated for 0, 12, 24, 36, 48, 72 h. The OD values at 450 nm wavelength for each well were measured using a microplate reader. The data was subsequently analyzed and processed to generate a proliferation curve.
Extraction of nuclear proteins
After co-culture with HIF cells for 48 h, NK92 cells were collected and lysed using a nuclear protein and cytoplasmic protein extraction kit (P0028, Beyotime) according to the manufacturer's guidelines.
RNA extraction, complementary DNA (cDNA) preparation and real-time reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from NK92 cells by TRIzol (15,596,018, Invitrogen), and cDNA was generated using 4 × EZscript Reverse Transcription Mix II (EZB-RT2G, EZBioscience, USA). The EZ-Probe qPCR Master Mix for microRNA (ROX2 plus) (EZB-miProbe-R2, EZBioscience) was used for real-time PCR. Each sample was analyzed in triplicate. The primer sequences used in this study are shown in Table 1.
Transcriptome analysis
HIF transfected with WNT2B plasmids or empty plasmids were co-cultured with NK92 cells for 24 h. The NK92 cells were subsequently collected and total RNA was extracted. RNA integrity was determined with an Agilent 2100 bioanalyzer. Next, mRNA was obtained, double-stranded cDNA was synthesized and end modified, fragments were selected and amplified by PCR to obtain the library, and then the libraries were detected. Different libraries were subjected to Illumina sequencing after pooling according to the effective concentration and the target data quantity. Functional enrichment analysis of differential genes, ingenuity pathway analysis (IPA), gene set enrichment analysis (GSEA), and protein interaction network analysis were then conducted.
Single cell sequencing analysis
Single cell data were downloaded from the GEO database. Cell normalization and regression were performed according to unique molecular identifier (UMI) count and mitochondrial percentage of each sample using Seurat software package (version 3.1.8). According to principal component analysis (PCA) of the first 2000 highly variable genes, the first 20 principal components were used for tSNE construction and Uniform Manifold Approximation and Projection (UMAP) construction. Clusters with the same cell types were selected for Re-tSNE analysis, graph-based clustering, and marker analysis. The marker genes of Seurat clustering results and the original expression count of the screened cells were selected. Intercellular communication molecules were analyzed systematically by CellPhoneDB. Relative activation of specific gene sets was then analyzed. Differentially expressed genes (DEGs) in the samples were identified and the gene co-regulatory network was determined. The Copy number variations\ (CNV) signal range of each cell was scored.
Western blotting
Cells were lysed with RIPA lysis buffer (P0013B, Beyotime) and protein concentrations in the cell lysates were quantified by Detergent Compatible Bradford Protein Assay Kit (P0006C, Beyotime). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to NC transfer membrane (HATF00010, Merck). Membranes were sealed with 5% skimmed milk powder and 2% BSA at room temperature for 1 h, incubated overnight with primary antibodies at 4℃, and then incubated with secondary antibodies at room temperature for 1 h. Finally, the membranes were visualized with SuperSignal™ West Pico PLUS (34,577, ThermoFisher Scientific).
Enzyme-linked immunosorbent assay (ELISA)
Cell culture supernatants were collected and the presence and concentration of wnt2b and IL-33 in the samples were determined following the steps described in the appropriate ELISA kit (SEL819Hu/SEB980Hu, Cloud-CloneCorp). Mouse colonic tissues were immersed in DMEM medium for 12 h, the supernatants were collected, and the concentration of IL-33 was determined using an ELISA kit (SEB980Mu, Cloud-CloneCorp) in accordance with the manufacturer's guidelines.
Co-immunoprecipitation (co-IP) analysis
Cell lysates were precleared by incubating with rabbit/mouse control IgG in Pierce™ Protein A/G Plus Agarose (80,423, Thermo) at 4℃ for 1 h. After centrifugation, the supernatant were incubated with primary antibodies at 4℃ overnight, and then with Pierce™ Protein A/G Plus Agarose at 4℃ for 2 h. Immunoprecipitates were collected and rinsed four times with cell lysis buffer. Sample were used to verify the interactome results by western blot.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) was performed with SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads) (9003, Cell Signaling Technologies) according to the manufacturer's instructions. NanoDrop (ThermoFisher Scientific) was used for DNA quantification, PCR was performed using the primers of IL33 promoter sequence (as shown in Table 1).
Hematoxylin and eosin (H&E)-staining and immunofluorescence
Colorectal segments of mice were removed, fixed in 4% formaldehyde for 48 h, embedded and sectioned (5 μm), and then used for H&E staining. For immunofluorescence (IF), sections were incubated with primary antibodies overnight at 4 °C, then incubated with fluorescence-labeled secondary antibodies at room temperature in the dark for 1 h, and DAPI was used to stain the nucleus.
Animal experiment
Cre mice were randomly divided into 2 groups. The modeled group was treated with rectal enema of TNBS (Sigma) and tail vein injection of AAV-NC, and it was referred to as "Control". The modeled group was treated with rectal enema of TNBS and tail vein injection of WNT2B KD Adeno-associated virus (AAV-WNT2B KD), and it was referred to as "WNT2B KD".Mice were injected with AAV(5 × 1010 physical particles of AAV in 200ul of PBS) via the tail vein at 28 days old, then the chronic TNBS models were constructed.
Induction of DSS-induced colitis and TNBS-induced colitis in mice.
For the acute DSS colitis model, 12 to 15-week-old pathogen-free male C57BL/6 J mice(27 ± 5 g) were numbered and weighed on the first day. The experimental group was injected with salinomycin (5 mg/kg) through abdominal cavity, while the control group was injected with physiological saline, and this regimen continued every other day for 7 days(days 1, 3, 5, 7). All mice received 2.5% DSS in drinking water on days 3, 5, and 7. Weight loss, stool consistency, and the degree of intestinal bleeding were recorded every day. All mice were sacrificed on day 10 and the colon and rectum were collected.
Chronic DSS and TNBS colitis models were constructed according to a previous study[18].
Masson staining
The paraffin sections were hydrated and stained with prepared Weigert's iron hematoxylin for 8 min, then differentiated in acid ethanol differentiation solution for 15 s, followed by water rinsing. Counterstained with Masson's blueing solution was performed for 5 min, followed by water rinsing. Distilled water rinse was performed for 1 min. Subsequently, the sections were stained with Ponceau acid fuchsin staining solution for 5 min, washed in weak acid working solution for 1 min, rinsed in phosphomolybdic acid solution for 1 min, and finally stained with aniline blue staining solution for 2 min following by a rinse in weak acid for 1 min.
Statistics
Results were expressed as mean ± SE. The software packages GraphPrism 8.0.2 and SPSS 15.0 were used for data analysis. Groups were compared by single factor analysis of variance (ANOVA) and then the minimum significant difference test was performed. Ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001 were considered significant.
Results
WNT2B high‑expressed fibroblasts and NK cells are enriched in colitis tissue
To determine whether WNT2B high‑expressed fibroblasts and NK cells are enriched in colonic tissue of IBD, the data of single cell sequencing for patients with IBD and healthy controls were extracted and analyzed[11, 19]. Among the main cell groups in colonic inflammatory tissues of patients with IBD, fibroblasts specifically expressing DCN, COL1A1, and PDGFRA accounted for a significant proportion of the total cells (Fig. 1A), and the WNT2B high‑expressed fibroblasts accounted for a large proportion of the fibroblast cluster (Fig. 1B). WNT2B high‑expressed fibroblasts enriched and upregulated immunoglobulin receptor binding and the humoral immune response in IBD (Fig. 1C). Among the immune cells, the proportion of NK cells specifically expressing NKG7 and KLRD1 in the main cell groups of colonic inflammation was similar to that of fibroblasts (Fig. 1D). The activated NK cells significantly enriched and regulated the canonical NF-κB pathway and altered immune responses (Fig. 1E). Fibroblasts-WNT2Bhi and NK cells in the colonic tissue of children with colitis, UC or CD were higher than in normal children, NK cells were significantly enriched in colon tissue of children with CD(Fig. 1F). In terms of cell interaction, WNT2B high‑expressed fibroblasts could act as upstream cells to interact directly with NK cells (Fig. 1G). Cellular immunofluorescence staining revealed that NK cells significantly expressed membrane receptor FZD4 (Fig. 1H). Finally, the number of NK cells specifically expressing FZD4 and CD56 and the number of fibroblasts expressing DPT in human colitis tissues were significantly higher compared with those in non-inflammatory colonic tissues (Fig. 1I-K). The infiltration of these two cell types in IBD inflammatory tissues showed a positive correlation (Fig. 1L). In summary, these data demonstrated that WNT2B high‑expressed fibroblasts and NK cells were enriched in the inflammatory colonic tissues.

WNT2Bhi fibroblasts and NK cells are enriched in colitis tissue. The transcriptome data of intestinal histiocytic cells (GSA:HRA000072) of PIBD and non-PIBD patients were analyzed. (A) tSNE plot displaying the main cell groups and fibroblasts in colonic tissue of PIBD patients. (B) tSNE plot displaying the proportion of WNT2Bhi fibroblasts in the fibroblast clusters. (C) WNT2Bhi fibroblasts enriched and upregulated the immune response. (D) tSNE plot displaying the main cell groups and NK cell groups in the colonic tissue of PIBD patients. (E) Activated NK cells in the colonic tissue of PIBD patients enriched and upregulated the immune response. (F) Fibroblasts-WNT2Bhi and NK cells in the colonic tissue of children with the corresponding disease. (G) CellPhoneDB analysis of other cells related to NK cells. (H) Immunofluorescence of FZD4 in NK cells. Magnification × 40; scale, 50 µm. (I)Immunofluorescence staining of NK cells expressing FZD4 and CD56 and fibroblasts expressing DPT in the inflammatory sites of PIBD patients. Magnification × 20 and × 40 respectively; scale, 100 µm and 50 µm, respectively. (J,K) Quantities of NK cells and fibroblasts in the immunofluorescence staining. (L) Relativity of NK cells and fibroblasts in the inflammatory sites of IBD patients
WNT2B high‑expressed fibroblasts promotes NK cells secreting IL-33
Given that WNT2B high‑expressed fibroblasts might connect with NK cells, and that wnt2b is secretory molecule, it was predicted that WNT2B high‑expressed fibroblasts might activate NK cells via wnt2b. To explore this hypothesis, wnt2b was overexpressed in colonic HIF to simulate the WNT2B high‑expressed fibroblasts (Fig. 2A). The concentration of wnt2b in the supernatants of WNT2B-overexpressing HIF was significantly higher compared with that of control fibroblasts (Fig. 2B). In addition, the proliferation activity of WNT2B high‑expressed fibroblasts was enhanced, and the proliferation activity began to show significant difference at 12 h(Fig. 2C). After co-culture with overexpressing and control HIF, a gene expression profile analysis was determined in the NK92 cells. This analysis showed that IFNA, IFNB, IL6, IL17, CXCL12, CXCR4, IL12, IL33, IL10, and IL10RA were the most differentially expressed immunity-related genes (Fig. 2D). The gene expression was further verified using RT-qPCR, and this revealed that only IFNA and IL33 were significantly higher expressed in NK92 cells co-cultured with WNT2B high‑expressed HIF (Fig. 2E). ELISA of the supernatant of NK92 cells showed that after exposure to WNT2B high‑expressed HIF, the NK92 cells secreted significantly increased amounts of IL-33 (Fig. 2F). There was no significant difference in IL-33 secretion between HIF cells with and without WNT2B overexpression(Figure S1A). Tissue immunofluorescence demonstrated that NK cells and IL-33 increased significantly in colonic tissue of mice with chronic colitis induced by TNBS and DSS (Fig. 2G). Furthermore, NK92 cells were pretreated with salinomycin (10 μM) for 12 h, an inhibitor of the WNT/β-catenin pathway, then co-culturing with different WNT2B-expressing HIF. Western blotting showed that while the co-culture with WNT2B high‑expressed HIF inhibited the phosphorylation of GSK3β and elevated the amount of β-catenin entering the nucleus of the NK cells, salinomycin inhibited this pathway, indicating that the WNT/β-catenin pathway in NK92 cells was activated by co-culturing with WNT2B high‑expressed HIF (Fig. 2H, I). Similarly, knocking down FZD4 induced a decrease in phosphor-GSK3β and the entry of β-catenin into the nucleus in NK92 cells, indicating that the WNT/β-catenin pathway was inhibited (Fig. 2J, K). Moreover, IL-33 expression levels showed the same trends as nucleus β-catenin (Fig. 2L), indicating that activation of the WNT/β-catenin pathway promoted IL-33 secretion.

WNT2Bhi fibroblasts promote secretion of IL-33 from NK cells. (A) Wnt2b expression from HIF transfection of WNT2B-GFP plasmid or empty plasmid were analyzed by Western blotting. (B) The concentration of wnt2b in supernatants of HIF transfection of WNT2B-GFP plasmid or empty plasmid. (C) CCK8 assay was used to detect the proliferation activity of HIF transfection of WNT2B-GFP plasmid or empty plasmid. (D) Whole gene expression profiling of differential gene expression in NK cells. Z-score > 0 indicated upregulation of gene expression, Z-score < 0 indicated downregulation of gene expression, Z-score ≥ 2 indicated significant upregulation of gene expression, and Z-score ≤ − 2 indicated significant downregulation of gene expression. (E) qPCR suggested the mRNA levels of DEGs in NK cells after co-culture with HIF transfected with WNT2B plasmid or empty plasmid. (F) The concentration of IL-33 in supernatants of NK cells co-cultured with HIF. (G) Infiltration of NK cells and IL-33 in colonic tissue of mice with chronic colitis induced by TNBS and DSS. Magnification × 40; scale, 50 µm. (H, I) NK92 cells were pretreated with salinomycin(10uM) or not for 12 h, then co-culturing with WNT2Bhi or wild-type HIF for 48 h. Expression of molecules involved in WNT/β-catenin pathway in NK92 cells analyzed by Western blotting. (J, K) NK92 cells transfected with control siRNA (siNC) or two different siRNA targeting FZD4 (siFZD4) were co-cultured with HIF after transfection of WNT2B-GFP plasmid or empty plasmid. Total proteins and nuclear proteins of NK92 cells were extracted and analyzed by Western blotting. (L) Quantification of the mRNA expression of IL-33 in NK cells, treated as (H). The differences were analyzed, and P values were calculated by GraphPrism8.0.2. *, P < 0.05; **, P < 0.01; ***, P < 0.001 were considered significant
Wnt2b activates NF-κB and STAT3 pathways
To explore the pathway activated by wnt2b, IPA was conducted for the whole gene expression profile of NK92 cells referenced in Fig. 2D. The DEGs were significantly enriched in inflammation-related pathways such as STAT3, PI3K/AKT, p38 MAPK, and NF-κB (Fig. 3A). To verify the activation of these pathways, NK cells were co-cultured with WNT2B high‑expressed or wild-type HIF and western blotting for members of these pathways was performed. Wnt2b markedly increased the phosphorylation of STAT3 (Tyr705) in NK92 cells, but there were no differences in proteins involved in the PI3K/AKT and p38 MAPK pathways (Fig. 3B-E). On this basis, salinomycin was shown to inhibit the phosphorylation of STAT3, IκBα, IKKα/β, and p65, and subsequently decrease the entry of STAT3 and p65 into the nucleus of NK cells (Fig. 3F, G). Cellular immunofluorescence and western blotting confirmed that wnt2b promoted entry of Stat3 and p65 into the nucleus of NK92 cells (Fig. 3H-J). Knocking down FZD4 expression also inhibited Stat3 phosphorylation and the NF-κB pathway, and consequently decreased entry of Stat3 and p65 into the nucleus (Fig. 3K, L). Together, these results suggest that wnt2b derived from fibroblasts binds to FZD4 and promotes activation of the STAT3 and NF-κB pathways.

Wnt2b activates NF-κB and STAT3 pathways. (A) Whole gene expression profile analysis showed the signaling pathways enriched by DEGs, the ratio was the number of differential genes divided by the number of whole genes contained in each pathway. (B-E) Expression of molecules involved in STAT, PI3K/AKT, p38 MAPK, and NF-κB pathways in NK cells whether the WNT/βcatenin pathway activated or not. (F) Protein expression of NF-κB pathways and phosphorylation of STAT3 in NK92 cells, and the quantified analysis of P-p65/p65 and P-Stat3/Stat3 in the three repeated WB experiments; the samples treatment as Fig. 2H. (G) STAT3 and p65 protein contents in the nucleus of NK92 cells, and the quantified analysis of p65 and Stat3 nuclear translocation in the three repeated WB experiments; the samples treatment as Fig. 2I. (H, I) Cellular immunofluorescence showed the entry of Stat3 and p65 into the nucleus of NK92 cells whether the WNT/βcatenin pathway was activated or not. Magnification × 40; scale, 50 µm. (J) Stat3 protein contents in the cytoplasm and nucleus of NK92 cells; the samples treatment as Fig. 3I. (K)Stat3 and p65 protein contents in the nucleus of NK92 cells, and the quantified analysis of Stat3 and p65 nuclear translocation in the three repeated WB experiments; the samples treatment as Fig. 2K. (L) Expression of molecules involved in NF-κB pathways and phosphorylation of STAT3 in NK92 cells, and the quantified analysis of P-Stat3/Stat3 and P-p65/p65 in the three repeated WB experiments; the samples treatment as Fig. 2J
Wnt2b promotes IL-33 expression by activating p65 and not STAT3
Since the NF-κB and STAT3 pathways were activated in NK cells co-cultured with WNT2B high‑expressed HIF, induction of the expression of IL-33 by these pathways was further investigated. A ChIP experiment was conducted using antibodies against p65 or STAT3, and revealed that wnt2b promoted p65, but not STAT3, binding to the IL-33 promoter (Fig. 4A-D). Knocking down either p65 or STAT3 inhibited their translocation into the nucleus (Fig. 4E,F), but only p65 knockdown reduced the expression of IL-33 (Fig. 4G). This indicated that wnt2b promotes IL-33 expression by activating p65, not STAT3.

Wnt2b promotes IL-33 expression by activating p65 instead of STAT3. (A) DNA electrophoresis of IL-33 promoter sequence binding to p65 performed by ChIP. (B) Gray values of DNA electrophoresis from Fig. 4A. (C) DNA electrophoresis of IL-33 promoter sequence binding to STAT3 performed by ChIP. (D) Gray values of DNA electrophoresis from Fig. 4C. (E, F) NK92 cells transfected with siNC or two different siRNA targeting p65 (siP65) or two different siRNA targeting STAT3(siSTAT3), were co-cultured with or without WNT2Bhi HIF, then total proteins and nuclear proteins of the NK92 cells were extracted and analyzed by Western blotting using the indicated antibodies. (G) NK92 cells transfected with siNC or two different siRNA targeting P65(sip65) or STAT3(siStat3) were co-cultured with HIF after transfection of WNT2B-GFP plasmid or empty plasmid for 24 h, and the total RNA of NK92 cells was extracted for qPCR analysis. P values were calculated by GraphPrism 8.0.2. *, P < 0.05; **, P < 0.01; ***, P < 0.001 were considered significant
TCF4 binds to p65 and upregulates the NF- κB pathway
TCF4 is a transcriptional factor located downstream of the WNT/β-catenin pathway. To explore the relationship between the WNT/β-catenin pathway and p65, TCF4 was knocked down in NK92 cells before co-culturing with WNT2B high‑expressed HIF. Activation of the WNT/β-catenin pathway led to the phosphorylation of IκBa, IKKa/b, and p65, representing activation of the NF-κB pathway (Fig. 5A). Since wnt2b induced p65 translocation into the nucleus, knocking down TCF4 obviously reduced the nuclear localization of p65 (Fig. 5B). To further verify the relationship between TCF4 and p65 location, mutated NLS variant of TCF4 was constructed, and it was found that TCF4's ability to promote the nuclear translocation of p65(Fig. 5C,D).Furthermore, co-immunoprecipitation showed that TCF4 bound to β-catenin and p65, and wnt2b promoted the binding of TCF4 and p65 (Fig. 5E-H). Immunofluorescence displayed that β-catenin, TCF4 and p65 were colocalized in the nucleus(Fig. 5I). These results indicated that β-catenin, TCF4 and P65 were combined into a ternary complex, while TCF4 bound to p65 and promoted the nuclear localization of p65.

TCF4 binds to p65 and upregulates the NF-κB pathway. (A, B) HIF transfected with WNT2B-GFP plasmid or empty plasmid were co-cultured with NK92 for 48 h; the NK92 cells were transfected with siNC or two different siRNA targeting TCF4(siTCF4). The total proteins and nuclear proteins of the NK92 cells were extracted and analyzed by Western blotting using the indicated antibodies, the quantified analysis of P-p65/p65 in the three repeated WB experiments of total proteins. NK cell transfection of MYC plasmid or TCF4-MYC plasmid or ΔNLS-TCF4-MYC plasmid were analyzed by Western blotting. Analysis of p65 in nuclear proteins located in NK cells by Western blotting, the samples treatment as Fig. 5C. (E–H) HIF transfected with WNT2B-GFP plasmid or empty plasmid were co-cultured with NK92 cells for 48 h. Total proteins of the NK92 cells were extracted and co-immunoprecipitation and Western blotting were performed with the indicated antibodies. (I) Immunofluorescence of β-catenin, TCF4, p65 in NK cells. Magnification × 40; scale, 50 µm
Salinomycin reduces IL-33 secretion in colitis
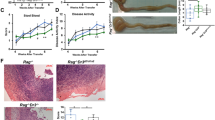
To determine whether inhibition of the WNT/β-catenin pathway has a protective effect on colitis, a series of indexes were measured in DSS-induced acute colitis model mice. Disease activity index scores showed that salinomycin significantly reduced colitis activity (Fig. 6A) and also significantly improved shortening of the colon and rectum, which are representative features of colitis (Fig. 6B-C). DSS-induced colitis mice exhibit increased infiltration of immune cells, destruction of the mucous membrane, disorder of the intestinal villus structure, crypt deformation, gland destruction, and abscess formation. Salinomycin significantly alleviated destruction of the mucous membrane and decreased the pathological scores of colonic inflammation, indicating that salinomycin reduced inflammatory responses (Fig. 6D-E). Detection of IL-33 in mice colonic tissues demonstrated that salinomycin reduced the secretion of IL-33 (Fig. 6F). Furthermore, immunofluorescence of mouse colonic tissues revealed that salinomycin reduced the expression of IL-33 in colonic inflammatory tissue (Fig. 6G). In summary, inhibition of the WNT/β-catenin pathway reduced IL-33 secretion in colonic inflammatory tissue and presented a protective effect on colitis.

Salinomycin reduces IL-33 secretion in colitis. In the process of acute enteritis induced by DSS, one group of mice were injected intraperitoneally with salinomycin (5 mg/kg), and the other group were injected with solvent as the Ctrl group..(A) Daily DAI changes of mice in Ctrl group and Salinomycin group. (B) Representative picture of colon of mice in each group. (C) Length of colon of mice in each group. (D) Representative pictures of H&E staining of colonic tissue of mice in each group. Magnification × 20 and × 50, respectively; scale, 100 µm and 40 µm, respectively. (E) Pathological score for colonic tissue of mice in each group. (F) The concentration of IL-33 in the supernatants of colonic tissue of Ctrl group and Salinomycin group. (G) Immunofluorescence of IL-33 in colonic tissue of acute DSS colitis mice treated with salinomycin or solvent. Magnification × 20 and × 40, respectively; scale, 100 µm and 50 µm, respectively. The differences were analyzed, and P values were calculated by GraphPrism 8.0.2. *, P < 0.05; **, P < 0.01; ***, P < 0.001 were considered significant
Knocking down WNT2B mitigates inflammation and fibrosis in chronic colitis
To investigate the role of WNT2B in chronic colitis, we employed a recombinant adeno-associated virus (AAV) vector-based gene delivery system as an innovative approach for knocking out WNT2B. B6.C-Tg(S100a4-cre) mice were administered AAV-WNT2B knockdown (AAV-WNT2B-KD) or control vectors (negative Adeno-associated virus, AAV-NC), followed by the construction of chronic colitis models using TNBS. The score of weight loss and stool condition showed that knocking down WNT2B reduced colitis activity (Fig. 7A) and also obviously improved shortening of the colon and rectum (Fig. 7B-C). There were obvious inflammatory cell infiltration and intestinal mucosal injury in TNBS-induced chronic colitis mice treated with AAV-NC. Knocking down WNT2B significantly alleviated destruction of the mucous membrane and decreased the pathological scores of colonic inflammation (Fig. 7D-E). Meanwhile, immunofluorescence of colonic tissues indicated that knocking down WNT2B mitigated the infiltration of NK cells and the expression of IL-33 in chronic colitis (Fig. 7F). in addition, it was found by Masson staining that knocking down WNT2B decreased the expression of fibrosis marker α-SMA in the intestinal tissues of chronic colitis(Fig. 7G). Immunofluorescence also confirmed the decreased expression of WNT2B and α-SMA in the intestinal tissues of chronic colitis with knocking down WNT2B (Fig. 7H). in conclusion, knocking down WNT2B inhibited NK cell infiltration and IL-33 secretion, mitigated inflammatory responses and fibrosis in chronic colitis.

Knocking down WNT2B reduces NK cell infiltration and IL-33 secretion in colitis, and reduce intestinal inflammation and fibrosis. In the process of chronic TNBS models, control group was treated with tail vein injection of AAV-NC, and the WNT2B KD group was treated with tail vein injection of WNT2B KD Adeno-associated virus (AAV-WNT2B KD). (A)The weight and stool condition were record in Control group and WNT2B-KD group. (B) Picture of colon of mice in each group. (C) Length of colon of mice in each group. (D)Representative pictures of H&E staining of colonic tissue of mice in each group. Magnification × 20 and × 40, respectively; scale, 100 µm and 50 µm, respectively. (E) Pathological score for colonic tissue of mice in each group. (F) Immunofluorescence of FZD4/NK1.1/IL-33 in colonic tissue of chronic TNBS and DSS colitis mice treated with AAV. Magnification × 20 and × 40, respectively; scale, 100 µm and 50 µm, respectively. (G) α-SMA was detected by Masson staining in each group of mice. (H) Immunofluorescence of α-SMA/WNT2B in colonic tissue of chronic TNBS colitis mice treated with AAV. Magnification × 20 and × 40, respectively; scale, 100 µm and 50 µm, respectively. The differences were analyzed, and P values were calculated by GraphPrism 8.0.2. *, P < 0.05; **, P < 0.01; ***, P < 0.001 were considered significant
Discussion
There is increasing evidence that fibroblasts involved in IBD by forming ECM and react directly or indirectly with immune cells [20]. Fibroblasts accomplish these effects by differentiating into different clusters such as myofibroblasts and inflammatory fibroblasts[21]. Besides these two main clusters, several more clusters of fibroblasts have been identified but their precise roles in IBD remain unclear[22]. Our previous research showed that there was a specific cluster of fibroblasts with high expression of WNT2B, and these WNT2B high‑expressed fibroblasts participated in not only inflammatory pathways such as the NF-κB and TNF pathways, but also the ECM pathway, indicating that this cell cluster had a dual phenotype[11]. In the current study, the gene expression patterns of WNT2B high‑expressed fibroblasts were reanalyzed and found to be enriched in B cell receptor signaling pathway and immune system process, indicating the WNT2B high‑expressed fibroblasts might take effect via immune cells. Moreover, CellPhoneDB analysis manifested that WNT2B high‑expressed fibroblasts interacted with NK cells directly as an upstream regulator, which has not previously been reported. NK cells are highly accumulated and play a pivotal role in IBD[15]. Moreover, the number of NK cells in the lamina propria of the colon decreased in patients treated with 6-mereaptopurine, which was associated with reduced disease activity[23, 24]. Results from the current study showed that both fibroblasts and NK cells were markedly accumulated in the inflammatory colonic tissues in IBD, and the infiltration of these two cell types showed a positive correlation, indicating that the WNT2B high‑expressed fibroblasts probably acted on NK cells.
Wnt2b belongs to the Wnt family, which has been reported as a secreted protein that binds to the FZD family and activates the classical WNT/β-catenin signal pathway[25]. FZD4 is the main receptor for WNT2B and is also significantly highly expressed in colonic tissues of IBD[26]. The classical WNT/β-catenin pathway is a highly conserved and tightly controlled signaling pathway involved in the regulation of embryonic development and cell proliferation and differentiation[27]. Activation of this pathway leads to nuclear translocation of β-catenin, and finally, the activation of target genes via TCF/LEF transcription factor[28]. Wnt2b-FZD4 interaction has been confirmed in peripheral eye development and enterocyte EMT in Crohn's disease through activation of the classical WNT/β-catenin pathway[29, 30]. The current study showed that NK cells markedly infiltrated the IBD colonic inflammatory tissue of both DSS- and TNBS-induced chronic colitis mouse models, and the NK cells significantly expressed FZD4. To further clarify the mechanism underlying these observations, we constructed the co-culture system of HIF and NK cells and determined that WNT2B high‑expressed fibroblasts secreting wnt2b activated the WNT/β-catenin pathway of NK cells via FZD4 and promoted IL-33 secretion from NK cells.
IL-33 belongs to the family of IL-1 cytokines and its receptor is suppression of tumorigenicity 2 (ST2). IL-33 is predominantly produced by epithelial cells, fibroblasts, and immune cells in the intestine, and participates in the progress of IBD[31, 32]. IL-33 can be secreted extracellularly as an inflammatory factor, acting on TH2 lymphocytes and mast cells, and regulating the phosphorylation of kinase (ERK), p38MAPK, and JNK and activation of the NF-κB signaling pathway[33, 34]. Recently, IL-33 was shown to be instrumental in IBD by promoting the TH1 response in Crohn’s disease[35, 36] and enhancing activation of the TH2 response related to ulcerative colitis[37]. In our research, IL-33 was demonstrated to be markedly elevated in colonic inflammation sites of the IBD-like colitis model, congruent with other studies. Furthermore, the study showed that secretion of wnt2b from WNT2B high‑expressed fibroblasts promotes the secretion of IL-33 from NK cells, and it can also promote the expression of IFNα.The expressions of IFN and IL-33 are increased in the tissues and serum of autoimmune diseases and partial infectious disease, which have demonstrated a positive correlation with disease severity[38]. In chronic pancreatitis, type I IFN recruits macrophage infiltrates and stimulates such macrophages to produce TNFα, which induces damaged pancreatic acinal cells to produce and release IL-33, while IFNAR knockout, the expression of IL-33 was inhibited, and it was found that IL-33 could effectively induce CD8 T cells to produce IFN-γ[39].In SLE, IL-33 up-regulates the expression of interferon regulatory Factor 7(IRF7) by acting on ST2 receptor, thereby inducing IFNα mRNA and protein expression[40]. This study also confirmed in animal experiments that knocking down WNT2B in intestinal tissue can reduce the expression of IL-33 and alleviate the fibrosis of intestinal tissue in chronic colitis.Since IL-33 is reported to promote the expression of fibrogenic factors IL-5, IL-13, and TGF-β1, leading to fibrosis of the GI tract in IBD[41, 42], findings from the current study provide a new insight that WNT2B high‑expressed fibroblasts might induce GI fibrosis via inflammatory-related pathway.
STAT3 is a member of STAT protein family. Phosphorylation of the tyrosine residue at Tyr705 of STAT3 protein leads to STAT3 nuclear translocation and downstream target gene transcription, while phosphorylation of Ser727 enhances the transcriptional activity[43]. In IBD, STAT3 activation promotes acute wound healing, but also leukocyte recruitment and the development of colitis-associated cancer during chronic inflammation[44]. The NF-κB pathway is a typical pro-inflammatory signaling pathway that promotes the expression of cytokines, chemokines, and adhesion molecules[45]. The WNT/β-catenin signal pathway could activate the NF-κB and STAT3 pathways[30, 46]. There is crosstalk and mutual activation between the NF-κB and STAT3 pathways. Inhibiting the STAT3 pathway can affect the NF-κB pathway. STAT3 can bind to the NF-κB complex containing IκB, displacing IκB from NF-κB, thereby promoting NF-κB activation and nuclear translocation[47]. Additionally, STAT3 may interact with p65 RelA/p65 in the nucleus and recruit the p300 histone acetyltransferase (HAT) complex to the complex[48]. Simultaneous activation of STAT3 and NF-κB can lead to their interaction with each other, further enhancing their activity. In common with these research findings, the current study showed that after activation of the WNT/β-catenin signal pathway, the NF-κB signaling pathway and STAT3 phosphorylation were both enhanced in NK cells, resulting in the translocation of p65 and STAT3 into the nucleus. However, since both p65 and STAT3 bound to the promoter sequence of IL-33, wnt2b selectively activated p65 to promote the expression of IL-33. This study also found that TCF4, a downstream transcription factor of WNT/β-catenin signaling, bound to p65 and promoted its nuclear translocation. The C-terminal of TCF4 contains a promoter-specific transactivation domain, which mediates the interaction with transcriptional coactivator p300 and activates the NF-κB pathway[49]. However, the current study provided the first evidence that TCF4 could directly bind to and activate p65, which might be a novel mechanism for WNT/β-catenin activation of the NF-κB pathway.
Salinomycin, an inhibitor of the WNT/β-catenin pathway, inhibits tumor growth by destroying the β-catenin/TCF complex[50]. Salinomycin is mainly used in tumor research. In the present study, salinomycin was used in the establishment of an acute DSS-induced IBD-like colitis model to explore the effect of the WNT/β-catenin signaling pathway on IBD. Salinomycin and knock down WNT2B decreased the colitis activity score[18] and colon tissue HE score[51], and increased the length of the colon and rectum, indicating protection from colonic inflammation. Furthermore, knock down WNT2B reduced the infiltration of NK cells and the secretion of IL-33. Therefore, our results provide the first evidence that salinomycin and knock down WNT2B have anti-inflammatory and anti-fibrotic effects and confirm the mechanism of WNT2B and WNT/β-catenin signaling inducing IL-33 secretion in vivo.
In summary, this study demonstrated that WNT2Bhi fibroblasts secreted wnt2b, activated the WNT/β-catenin pathway of NK cells via FZD4, and promoted NF-κB signaling and IL-33 expression (Fig. 8), further aggravating intestinal injury and inducing intestinal fibrosis. Findings from the study may facilitate the establishment of new therapeutic approaches to prevent IBD complications.

Schematic illustration for the crosstalk of WNT2Bhi fibroblasts and NK cells in IBD. Wnt2b binds to FZD4 to activate the WNT/β-catenin pathway. WNT/β-catenin pathway mediates phosphorylation of p65 and stat3, leading to increase p65 and stat3 nuclear translocation. Whereas p65 binds to the promoter of IL-33 to promote the expression of IL-33. Importantly, TCF4 interacts with p65 and regulates NF-κB pathway
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Abbreviations
- FZD4:
-
Frizzled class receptor 4
- DSS:
-
Dextran sulfate sodium;
- TNBS:
-
2,4,6-Trinitrobenzene sulfonic acid
- IBD:
-
Inflammatory bowel disease
- DCN:
-
Decorin
- COL1A1:
-
Collagen type I alpha 1 chain
- PDGFRA:
-
Platelet-derived growth factor receptor alpha
- FOSB:
-
FosB proto-oncogene, AP-1 transcription factor subunit
- NKG7:
-
Natural killer cell granule protein 7
- KLRD1:
-
Killer cell lectin like receptor D1
References
Guan Q (2019) A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J Immunol Res 2019:7247238
Ramos GP, Papadakis KA (2019) Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin Proc 94(1):155–165
Zhang M, Zhang S (2020) T Cells in Fibrosis and Fibrotic Diseases. Front Immunol 11:1142
Latella G et al (2015) Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol 50(1):53–65
Powell DW et al (2011) Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol 73(1):213–237
Vetuschi A et al (2018) PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur Rev Med Pharmacol Sci 22(24):8839–8848
Wang J et al (2021) Novel mechanisms and clinical trial endpoints in intestinal fibrosis. Immunol Rev 302(1):211–227
Cook SA, Schafer S (2020) Hiding in Plain Sight: Interleukin-11 Emerges as a Master Regulator of Fibrosis, Tissue Integrity, and Stromal Inflammation. Annu Rev Med 71(1):263–276
Zhang F et al (2019) Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol 20(7):928–942
Wei K, Nguyen HN, Brenner MB (2021) Fibroblast pathology in inflammatory diseases. J Clin Invest 131(20):e149538
Huang B et al (2019) Mucosal Profiling of Pediatric-Onset Colitis and IBD Reveals Common Pathogenics and Therapeutic Pathways. Cell 179(5):1160-1176.e24
Mack M (2018) Inflammation and fibrosis. Matrix Biol 68–69:106–121
Sanos SL, Diefenbach A (2010) Isolation of NK cells and NK-like cells from the intestinal lamina propria. Methods Mol Biol 612:505–517
Zaiatz BV et al (2021) Dysregulation of Metabolic Pathways in Circulating Natural Killer Cells Isolated from Inflammatory Bowel Disease Patients. J Crohns Colitis 15(8):1316–1325
Poggi A et al (2019) Human Gut-Associated Natural Killer Cells in Health and Disease. Front Immunol 10:961
Balsamo M et al (2009) Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A 106(49):20847–20852
Greb-Markiewicz B et al (2019) The subcellular localization of bHLH transcription factor TCF4 is mediated by multiple nuclear localization and nuclear export signals. Sci Rep 9(1):15629
Wirtz S et al (2017) Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc 12(7):1295–1309
Martin JC et al (2019) Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 178(6):1493-1508.e20
Barnhoorn MC et al (2020) Stromal Cells in the Pathogenesis of Inflammatory Bowel Disease. J Crohns Colitis 14(7):995–1009
Nowarski R, Jackson R, Flavell RA (2017) The Stromal Intervention: Regulation of Immunity and Inflammation at the Epithelial-Mesenchymal Barrier. Cell 168(3):362–375
Kinchen J et al (2018) Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 175(2):372-386.e17
Yusung S et al (2017) NK cells are biologic and biochemical targets of 6-mercaptopurine in Crohn’s disease patients. Clin Immunol 175:82–90
Steel AW et al (2011) Increased proportion of CD16(+) NK cells in the colonic lamina propria of inflammatory bowel disease patients, but not after azathioprine treatment. Aliment Pharmacol Ther 33(1):115–126
Nusse R (2012) Wnt signaling. Cold Spring Harb Perspect Biol 4(5):a011163
You J et al (2008) Wnt pathway-related gene expression in inflammatory bowel disease. Dig Dis Sci 53(4):1013–1019
He S, Tang S (2020) WNT/β-catenin signaling in the development of liver cancers. Biomed Pharmacother 132:110851
Liu J et al (2022) Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther 7(1):3
Ohta K et al (2011) Tsukushi functions as a Wnt signaling inhibitor by competing with Wnt2b for binding to transmembrane protein Frizzled4. Proc Natl Acad Sci U S A 108(36):14962–14967
Ortiz-Masià D et al (2020) WNT2b Activates Epithelial-mesenchymal Transition Through FZD4: Relevance in Penetrating Crohn´s Disease. J Crohns Colitis 14(2):230–239
Hodzic Z et al (2017) IL-33 and the intestine: The good, the bad, and the inflammatory. Cytokine 100:1–10
Kurimoto M et al (2021) IL-33 as a Critical Cytokine for Inflammation and Fibrosis in Inflammatory Bowel Diseases and Pancreatitis. Front Physiol 12:781012
Kakkar R, Lee RT (2008) The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov 7(10):827–840
Schmitz J et al (2005) IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23(5):479–490
De Salvo C et al (2021) NOD2 drives early IL-33-dependent expansion of group 2 innate lymphoid cells during Crohn's disease-like ileitis. J Clin Invest 131(5):e140624
Baumann C et al (2015) T-bet- and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc Natl Acad Sci U S A 112(13):4056–4061
Pastorelli L et al (2010) Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A 107(17):8017–8022
Minaga K et al (2020) Identification of serum IFN-α and IL-33 as novel biomarkers for type 1 autoimmune pancreatitis and IgG4-related disease. Sci Rep 10(1):14879
Watanabe T et al (2016) Nucleotide-binding oligomerization domain 1 acts in concert with the cholecystokinin receptor agonist, cerulein, to induce IL-33-dependent chronic pancreatitis. Mucosal Immunol 9(5):1234–1249
Georgakis S et al (2021) NETs decorated with bioactive IL-33 infiltrate inflamed tissues and induce IFN-α production in patients with SLE. JCI Insight 6(21):e147671
Liew FY, Girard JP, Turnquist HR (2016) Interleukin-33 in health and disease. Nat Rev Immunol 16(11):676–689
Sedhom MA et al (2013) Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 62(12):1714–1723
Wang Y et al (2018) The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett 415:117–128
Willson TA et al (2012) STAT3 genotypic variation and cellular STAT3 activation and colon leukocyte recruitment in pediatric Crohn disease. J Pediatr Gastroenterol Nutr 55(1):32–43
Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1(6):a001651
Guan X et al (2021) Crosstalk between Wnt/β-catenin signaling and NF-κB signaling contributes to apical periodontitis. Int Immunopharmacol 98:107843
Fan Y, Mao R, Yang J (2013) NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 4(3):176–185
Grivennikov SI, Karin M (2010) Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 21(1):11–19
Hecht A, Stemmler MP (2003) Identification of a promoter-specific transcriptional activation domain at the C terminus of the Wnt effector protein T-cell factor 4. J Biol Chem 278(6):3776–3785
Wang Z et al (2019) Salinomycin exerts anti-colorectal cancer activity by targeting the β-catenin/T-cell factor complex. Br J Pharmacol 176(17):3390–3406
Neurath MF et al (1995) Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med 182(5):1281–1290
Acknowledgements
We thank Dr. Hong Du and Dr. Yongqiang Li of Guangzhou First People's Hospital for providing IBD tissue samples.
Funding
This work was supported by Construction project of Guangzhou Medical Key Subject (Pediatric Department of Gastroenterology) (2021–2023), Guangzhou Municipal Health Commission (grant number 011006003), National Natural Science Foundation of China (No. 82073174 and No. 81802339), Foundation for Basic and Applied Basic Research of Guangdong Province (grant number 2022A1515012406), Foundation for Basic and Applied Basic Research of Guangzhou (grant number 202102020185).
Author information
Authors and Affiliations
Contributions
Yanling Cheng, Shuzhe Xiao, Lanlan Geng, Yang Cheng, and Sitang Gong conceived the experiments and analyzed the data. Yanling Cheng, Yang Cheng, and Sitang Gong wrote the manuscript. Yanling Cheng, Shuzhe Xiao, Lin Lan, Yun Zhu, Danqiong Liu, Rui Tang, Zhihua He, and Jianbiao Gu conducted the experiments. Shuzhe Xiao and Lanlan Geng provided human IBD tissue samples. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethical Approval
Human study: Based on the declaration of Helsinki as reflected in a prior approval by the institution's human research committee, this study was approved by Guangzhou Women and Children's Medical Center ethics committee. Animal study: All animal experiments were approved by institutional Animal Experiment Committee of Southern Medical University or Nanfang Hospital, and performed at Nanfang Hospital.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest to disclose.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
Cite this article
Cheng, Y., Xiao, S., Lan, L. et al. WNT2B high‑expressed fibroblasts induce the fibrosis of IBD by promoting NK cells secreting IL-33. J Mol Med (2024). https://doi.org/10.1007/s00109-024-02477-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00109-024-02477-x