
Abstract
This paper reviews the current status of our understanding of the epidemiology, diagnosis, and management of the continuum of pancreatic diseases from acute and recurrent acute pancreatitis to chronic pancreatitis and the diseases that are often linked with pancreatitis including diabetes mellitus and pancreatic cancer. In addition to reviewing the current state of the field, we identify gaps in knowledge that are necessary to address to improve patient outcomes in these conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview
Chronic pancreatitis (CP) involves progressive inflammatory and fibrotic changes of the exocrine pancreas resulting in permanent structural damage, which can lead to impairment of both endocrine and exocrine functions. The epidemiological data on CP is not well defined due to difficulty in early diagnosis and the variable clinical progression from acute to CP. However, the current literature points to a rising incidence and prevalence of CP. A population-based study from Mayo Clinic found increased incidence from 2.94/100,000 during 1977–1986 to 4.35/100,000 during 1997–2006, with a prevalence rate of 41.76 per 100,000 populations in Olmsted County, MN [1]. A retrospective study from 1996 to 2003 in China found the prevalence of CP rose from 3.08 to 13.52 per 100,000 [2]. Two parts of India have reported a prevalence of 20–125/100,000 persons for tropical CP [3, 4]. Despite the relatively low prevalence of CP, the disease costs the US healthcare system over 150 million dollars yearly [5].
While acute pancreatitis (AP) affects both men and women equally, CP is found to be more common in men [6]. Blacks also have been shown to have a two-to-threefold greater prevalence of CP [7].
Clinical manifestations include mid-epigastric abdominal pain with radiation to the back, worsened with food, and associated with nausea and vomiting. As the disease progresses, the ongoing loss of pancreatic exocrine function can lead the pancreas to “burn itself out,” resulting in a decrease or complete resolution of pain [8]; however, this topic remains highly controversial. Fat malabsorption and steatorrhea can occur due to pancreatic exocrine dysfunction [9]. Pancreatic endocrine insufficiency can also occur, leading to glucose intolerance and ultimately diabetes mellitus [10]. Diabetes occurring secondary to exocrine pancreatic disease is categorized as type 3c diabetes mellitus (T3cDM) [11]. Similar to type 1 diabetes, T3cDM is insulin dependent, but different due to the involvement of α cells, which impedes glucagon production, increasing the risk of hypoglycemia [9]. Moreover, T3cDM rarely develop diabetic ketoacidosis because there is not complete loss of beta cell function [12].
CP has been found to be associated with a nearly 50% mortality rate within 20–25 years of diagnosis [13, 14] due to factors including infection, malnutrition, and complications from recurrent pancreatitis. Additionally, CP is the strongest identified risk factor for pancreatic cancer and increases the risk at least 13.3-fold [6]. Moreover, patients with both CP and diabetes have a 33-fold increased risk of pancreatic cancer [11].
Given the rise in incidence and prevalence of CP, the potential complications and high mortality rate, it is imperative that physicians understand the risk factors, disease process, and management of this disease. Importantly, a better understanding of the mechanism behind CP is necessary in order to develop therapeutic options to prevent the progression of CP and the development of T3cDM and pancreatic cancer.
Recurrent Acute Pancreatitis and Chronic Pancreatitis
In 1946, Comfort et al. [15] first proposed the theory that recurrent acute pancreatitis (RAP) can lead to CP based on their histological findings of AP in patients with CP. However, the Marseilles Criteria and the revised Marseilles Criteria in the 1980s suggested that AP and CP are two distinct diseases [16].
Our current literature has trended back to the initial proposal that RAP can lead to CP. Two studies reported progression from AP to CP varying from 4 to 24% [17, 18]. Yadav et al. reported progression from AP to CP in 12.8% of patients, and found that RAP was the strongest predicting factor for a subsequent diagnosis of CP. In this study, alcohol and tobacco abuse were leading independent predictors for RAP [19]. Furthermore, patients with hereditary pancreatitis have recurrent episodes of AP that can ultimately lead to CP [20].
The necrosis–fibrosis theory supports the notion of RAP as an etiology of CP [21]. Inflammation and necrosis from repeated episodes of AP lead to scarring and marked fibrosis with ductal obstruction [15, 22]. Additionally, the Sentinel Acute Pancreatitis Event (SAPE) Hypothesis theorized that the first episode of AP (sentinel event) sensitizes the pancreas to permanent fibrosis and each subsequent pancreatitis episode leads to more inflammation and fibrosis resulting in loss of glandular structure and function [23].
Environmental Factors Associated with Recurrent and Chronic Pancreatitis
Specific risk factors for CP include environmental factors such as alcohol and smoking, genetics, and obstructive diseases. In some patients, etiologies of CP are never found.
Alcohol
Alcohol is thought to be the leading cause of CP. In the USA, alcohol has been found to be the etiology in nearly 50% of cases of CP [2]. Following an episode of alcohol-related AP, the risk of progression to CP was approximately 14% with complete abstinence or occasional drinking, 23% with decreased but daily drinking daily, and 41% with no change in drinking [24]. Yadav et al. [25] found the threshold of five drinks or more per day as an increased risk for CP. Moreover, multiple meta-analyses revealed that increased alcohol consumption exponentially increased the risk of CP [26,27,28].
However, a recent study found that moderate alcohol intake (less than 2 drinks per day) was protective against recurrent acute and CP [29]. This finding was verified in animal models where ethanol feeding inhibits the activation of nuclear factor-κB, a pro-inflammatory transcription factor, in the pancreas, and upregulates a protective endoplasmic reticulum stress response [30,31,32,33,34].
Interestingly, only 3% of alcoholics develop CP, suggesting other risk factors may play a key role in supplementing alcohol effects in disease progression [35]. Animal models suggest ethanol increases the risk of pancreatitis in the setting of a second risk factor [30], such as smoking [29].
Genetics has also been found as a second risk factor in alcoholics. Whitcomb et al. [36] discovered an association between genetic variants of CLDN2 in alcoholic patients. CLDN2 is an X-linked gene that encodes the protein Claudin-2, which is highly expressed by pancreatic acinar cells during stressful conditions, and may contribute to pathologic inflammation of CP [37].
Smoking
Smoking and drinking are common co-existing behaviors and synergistically may contribute to the development of CP. A study of 108 smokers with alcohol-related CP found that smoking accelerates the progression of pancreatic disease in a dose-dependent fashion, distinct from the level of alcohol consumption [25].
Smoking has also been found to be an independent risk factor for CP. A meta-analysis conducted in 2010 found the pooled risk estimates for smoking was 2.5 (95% CI 1.3–4.6) after adjusting for alcohol consumption. Smoking increased the risk of CP in a dose-dependent relationship with a twofold increase in risk of smoking less than one pack per day and more than threefold increase risk of smoking one or more packs per day [38]. For former smokers, the relative risk estimate dropped to 1.4 (95% CI 1.1–1.9) [38]. Furthermore, smoking not only increases the risk for CP, but also increases the risk for pancreatic cancer with a relative risk of 15.6 (95% CI 7.48–28.7) for smokers compared to non-smokers [39]. Therefore, smoking cessation may prove an important therapeutic intervention as it may decrease the risk of both CP and pancreatic cancer.
Genetic Factors
Hereditary pancreatitis (HP) is associated with RAP and CP. The diagnosis of HP is made by genetic testing but can be supported by clinical and family history. HP was first described in six family members spanning three generations in 1952 [20]. All family members were diagnosed with early onset pancreatitis before the third decade of life and had chronic recurrent pancreatitis. A history of acute recurrent pancreatitis during childhood and family history of recurrent/CP in two first-degree relatives or in three second-degree relatives should raise the suspicion for possible HP [40].
Since the first report, more literature has emerged with the identification of multiple genes implicated in disease development. The first genetic defect was discovered in 1996. A gain-of-function mutation in the PRSS1 gene, which codes for trypsin, was found to cause HP [41]. Normally, trypsin converts inactive pancreatic zymogens into active digestive enzymes in the duodenum. However, premature conversion of trypsinogen to trypsin leads to premature activation of pancreatic zymogens in the pancreas; ultimately, pancreatic parenchymal damage and pancreatic fibrosis occur leading to RAP/CP. Inheritance occurs as an autosomal dominant trait with variable expression [42].
CP has also been associated with loss-of-function mutations. The SPINK1 and CTRC genes encode for two different proteins that both inhibit trypsin. Thus, loss of SPINK1 and CTRC can lead to auto-digestion and pancreatitis [43, 44].
Mutations in the cystic fibrosis transmembrane regulator (CFTR) are also associated with CP. CFTR is critical for the secretory function of the exocrine pancreatic duct cells to promote the flow of digestive enzymes into the duodenum preventing pancreatitis [45]. Mutations in CFTR have been shown to be associated with CP without pulmonary manifestations of cystic fibrosis [46]. Interestingly, recent studies have shown that alcohol abuse inhibits CFTR function supporting a crucial role for ductal function in preventing pancreatitis [47].
Anatomic and Obstructive Abnormalities
Ductal obstruction secondary to inflammatory strictures or malignancies can lead to chronic obstructive pancreatitis. Pancreas divisum can lead to RAP and subsequent CP [48]. A higher frequency of pancreas divisum has been seen in patients with CFTR mutation [49], suggesting pancreas divisum may be acting synergistically with genetic factors.
Other Factors in Recurrent and Chronic Pancreatitis
Despite many different etiologies for CP, 10–30% patients have no identifiable causative factor in a bimodal distribution [21, 50, 51]. Possible mechanisms for early and late onset idiopathic CP include undiagnosed genetic defects and occult alcohol use. A form of idiopathic early onset CP is tropical pancreatitis, also known as fibrocalculous pancreatic diabetes. It is found in tropical regions of the world with the higher prevalence in Southern India at 20–125/100,000 persons [3, 4].
Mechanisms and Potential of Therapeutics Development
CP occurs as a result of sustained chronic inflammation and fibrosis of the pancreas. Understanding these processes on a cellular and molecular level is important to create future therapies in hopes of preventing the progression of RAP to CP.
During episodes of AP, the parenchymal cells (acinar and ductal) cells produce pro-inflammatory cytokines, which recruit inflammatory cells and lead to further injury and potential tissue necrosis. The propagation of the acute inflammatory response can lead to chronic inflammation if there is not appropriate resolution [30, 52,53,54,55,56]. On a molecular level, pancreatic stellate cells (PaSCs) have been found to have a definite linkage in models of CP [57,58,59,60]. PaSCs are normally present in a “quiescent” state in the exocrine pancreas surrounding the acinar and ductal structures and providing the basement membrane structure and organization of the pancreatic epithelium [61]. However, in CP, PaSCs participate in disease pathogenesis after transforming into an activated or “myofibroblastic” state [61]. In this myofibroblastic state, PaSCs produce collagen and other extracellular matrix proteins that lead to fibrosis; moreover, PaSCs secrete cytokines that further promote the inflammatory process [57, 62].
Tumor growth factor beta (TGF-β) is a cytokine that has been shown to play a key role in fibrosis development through the activation of PaSCs [57, 59, 63, 64]. Additionally, animal models propose that the mechanism of disease progression is due to a feed-forward interaction between the PaSC and a key inflammatory cell, the alternatively activated macrophage [65]. Alternatively activated macrophages secrete TGF-β, which maintains PaSCs in the myofibroblastic state, thereby promoting inflammation and fibrosis. In turn, TGF-β-stimulated PaSCs produce key cytokines such as interleukin (IL)-4 and IL-13, which promote the alternatively activated state of the macrophages. This feed-forward promotion is necessary for inflammation and fibrosis.
Interestingly, a recent paper showed that smoking, through interaction with the aryl hydrocarbon receptor on T cells, also stimulated PaSCs to promote fibrosis through the IL-22-pathway [66]. These findings again reveal an important interplay between inflammatory cells and PaSCs in CP pathogenesis.
This discussion points out that therapies that halt acute inflammation and prevent recurrent episodes as well as those directed to the PaSC and its interactions with the immune system will play a central role in preventing and curing chronic pancreatitis.
Diagnosis of Chronic Pancreatitis
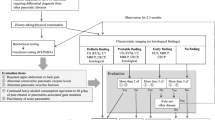
The diagnosis of CP is based on a combination of clinical history, risk factors, imaging, endoscopy, and pancreatic function testing. Currently, the early diagnosis of CP is difficult due to a lack of sensitive blood, imaging and functional biomarkers.
Blood Tests
In AP, amylase and lipase are typically elevated, whereas in CP, the serum concentrations of these enzymes are usually normal to mildly elevated due to loss of functional exocrine pancreatic tissue from pancreatic fibrosis [67]. The white cell count and electrolytes are usually unremarkable, unless diminished intake, vomiting, or digestive insufficiency has occurred. Elevations of serum bilirubin and alkaline phosphatase can occur, which suggests compression of the intrapancreatic portion of the bile duct by edema, fibrosis, or pancreatic cancer [9].
Circulating biomarkers associated with CP are not well established. Two recent studies have shown increased level of TGF-β. Other studies have reported increases in matrix metalloproteinase-9 (MMP-9) [68], tumor necrosis factor-α (TNF-α) [68], and soluble fractalkine [69]. Interestingly, IL-22 has also shown to be increased in CP patients who are smokers [66]. Thus, more studies are needed to identify, verify, and validate novel clinical biomarkers of disease activity to aid in early diagnosis of CP.
Imaging
Imaging can play a key role in diagnosing CP. Various imaging modalities including transabdominal ultrasound, endoscopic ultrasound (EUS), computerized tomography (CT) scan, and magnetic resonance imaging (MRI) can be used to detect morphological changes in the pancreas. Pancreatic calcifications are pathognomonic for severe CP and are located exclusively in the ductal system [67]. However, diagnosing CP from imaging alone is challenging given that morphologic changes may not appear on imaging until later in the disease. This presents a challenge in diagnosing those with early mild or moderate disease.
Transabdominal ultrasound and CT imaging can be used to detect advanced disease. While ultrasound is relatively inexpensive and free of radiation, its ability to visualize the pancreas is poor compared to other imaging modalities. Multiple echogenic foci representing calcifications are the classic findings seen on ultrasound. These are seen in only up to 40% of patients [70]. CT imaging has been shown to have sensitivity ranging from 74 to 90% and a specificity of 80–90% in diagnosing advanced CP [71]. Common findings on CT imaging include pancreatic ductal dilatation, parenchymal atrophy, and pancreatic calcifications [71]. CT imaging is considered to be the best initial imaging test for CP because of its high sensitivity and specificity, and its ability to potentially identify other causes of abdominal pain [72].
Magnetic resonance cholangiopancreatography (MRCP) and MRI have also been used to diagnose CP and have the advantage of no radiation exposure. Moreover, MRI has the advantage of detecting both parenchymal and ductal changes [73]. MRI/MRCP can be combined with hormonal stimulation using intravenous secretin to aid in the diagnosis of early CP with a sensitivity of 77% and specificity of 83% [72, 74].
EUS has emerged as an important imaging modality to detect early morphologic changes in CP. It can detect mild parenchymal and ductal changes not seen on CT scan, and can be used when CT and MR imaging are non-diagnostic [75, 76]. There are nine criteria used in diagnosing CP with EUS: four parenchymal features including hyperechoic foci, hyperechoic strands, lobular contour, and cysts, and five ductal features including main duct dilatation, duct irregularity, hyperechoic margins, visible side branches, and stones [72]. Currently there is no firmly established number of criteria needed to diagnose CP, but the sensitivity and specificity increases with increasing number of criteria [77]. One study showed sensitivity and specificity of 83 and 80%, respectively, when using at least three criteria [78], and another study showed sensitivity and specificity of 84 and 100%, respectively, when using at least four EUS criteria [79]. EUS quantitative elastography can also be used to quantify the degree of fibrosis and help stage the degree of CP. Iglesias et al. [80,81,82] found a strain ratio cutoff point of 2.25 to have diagnostic sensitivity and specificity of 91% for CP and found good correlation with EUS criteria.
One of the greatest limitations in using EUS is the low interobserver agreement. Studies show good agreement in two features, duct dilatation (κ = 0.6) and lobularity (κ = 0.51), but low agreement for the other seven features (κ < 0.4) [83].
Endoscopic retrograde cholangiopancreatography (ERCP) was considered to be the gold standard to detect early changes. However, this procedure is invasive, expensive, and time consuming. In addition, ERCP can only evaluate for ductal changes. Moreover, given the advent of MRCP and EUS, ERCP has less of a role in diagnosing CP. The most recent guidelines by the American Society for Gastrointestinal Endoscopy (ASGE) in 2006 recommended reserving the use of ERCP for patients in whom diagnosis is inconclusive despite pancreatic function testing CT/MRI or EUS [84]. The Cambridge Criteria is used to diagnose CP by ERCP, which determines normal or equivocal to mild, moderate, or severe CP based on main duct and side branch abnormalities [85]. Studies evaluating the accuracy of ERCP findings were compared to histopathology findings in patients with CP, and found that for patients in the early disease group (normal, equivocal, mild based on the Cambridge Criteria), ERCP findings correlated with histopathology findings in 67% of patients and 77% in those with moderate and severe disease [86].
Considering this background, there is general consensus that EUS represents the current most sensitive imaging test based on subsequent histo-pathological examination in surgical specimens [87,88,89]. However, the role of EUS in identifying patients with non-calcific CP is controversial. One study showed EUS has good efficacy in diagnosing early non-calcific CP with sensitivity and specificity of 90.5 and 85.7%, respectively, when compared to histopathology using at least four EUS criteria [87]. However, in another study examining patients with abdominal pain and non-calcific CP requiring total pancreatectomy and islet auto-transplantation, sensitivity of 61% was found [90]. Another caveat is that both EUS findings of CP as well as histopathologic features of CP are found in several conditions without symptomatic clinical presentations. These include age (i.e., over 60 years old) [89, 91], ethanol effects, and lifestyle factors [92,93,94]. The frequency of EUS detected abnormalities in patients with no clinical evidence of CP increases with age especially in those over 60 years [91], and it is unclear whether these findings have clinical significance [89].
Thus, the use of EUS for diagnosis has some limitations and should be interpreted in the clinical context. Furthermore, advances in imaging methods are needed to aid with diagnosis and management.
Pancreatic Function Testing
The role of pancreatic function testing is limited based on practicality in comparison with the ease of imaging modalities as previously discussed. However, functional testing can be considered in cases with equivocal morphological imaging [95].
End-stage CP occurs when more than 90% of exocrine pancreatic function is lost and ultimately leads to pancreatic exocrine insufficiency (PEI) and steatorrhea [96]. A 72-h quantitative fecal fat determination can be used to diagnose steatorrhea, though it is not specific for CP and can be seen in small bowel mucosal disease such as celiac disease, Crohn’s disease, and bacterial overgrowth [97].
More specific fecal tests for PEI include fecal chymotrypsin and elastase-1. Both enzymes are produced by the pancreas and remain constant throughout the gastrointestinal tract. Elastase-1 has been shown to be more specific than chymotrypsin with sensitivity approaching 100% for severe insufficiency, and specificity reported as 93% [98, 99]. While this test may be good for patients with severe CP, those with early or mild CP have been found to be less sensitive [100].
PEI can also be diagnosed with serology through the measurement of trypsinogen. Trypsinogen reflects pancreatic acinar mass, and levels of serum trypsinogen below 20 ng/ml were found to have a high sensitivity for severe PEI [101].
Breath tests have also been developed to evaluate pancreatic exocrine function. Patients ingest 13C-marked substrates with a test meal, which is then hydrolyzed in the duodenum in proportion to the amount of pancreatic exocrine function. The hydrolyzed products are absorbed, metabolized, and will eventually reach the pulmonary endothelium where it is released with expiration [102]. There are many breath tests that can measure pancreatic function, but the most sensitive test involves assessing lipase activity as it has been shown to be the first enzyme impaired in pancreatic insufficiency [103]. There are many different test meals to assess lipase activity, all centering on a meal with high triglyceride content. The most investigated is a mixed triglyceride breath test, which found a sensitivity of 89% and specificity of 81% for diagnosis of pancreatic steatorrhea [104]. While it has good sensitivity for advanced disease, Loser et al. [105] showed a sensitivity of 46% for mild disease when compared to the secretin-caerulein test.
Direct testing of pancreatic function by measuring secretions from the exocrine pancreas has a higher sensitivity for CP when compared to the methods described above. Direct pancreatic function tests involve direct stimulation of the pancreatic duct and acinar cells using secretagogues. While this method is more invasive and time consuming compared to the indirect method, it is more accurate in diagnosing early CP [102]. There are many different types of direct pancreatic function testing. The Lundh test is considered to be the most physiologic, but it is no longer used clinically as the sensitivity was low compared to hormone-stimulated tests consisting of secretin or cholecystokinin stimulation [106].
While both pancreatic acinar and ductal cells are compromised in severe CP, currently testing the ductal function with secretin stimulation prevails over testing acinar function with cholecystokinin stimulation [107]. In a retrospective study performed in 2013, Ketwaroo et al. [108] examined patients with suspected CP but with normal imaging studies who underwent secretin pancreatic function testing, and the sensitivity and specificity were found to be 82 and 86%, respectively. Furthermore, the negative predictive value was found to be 97% [108]. Therefore, in patients with suspected early CP with negative imaging, secretin pancreatic function testing may be useful in providing evidence supporting the diagnosis.
There are currently no clinically validated and utilized markers of fibrosis and inflammation in pancreatic fluid although preliminary studies have demonstrated that this fluid can be used for a variety of measures related to inflammation and fibrosis [109]. In near future, it may be possible that studies of pancreatic fluid can lead to biomarkers of inflammation and fibrosis that characterize subsets of patients and response to specific therapeutic interventions.
Management of Chronic Pancreatitis
Abdominal pain is the most debilitating symptom in patients with CP. Thus, most therapies are centered on alleviating abdominal pain. Increasing evidence has shown that progressive development of fibrosis and subsequent loss of normal pancreatic tissue and ductal patency and secretion, along with chronic inflammation involving intrapancreatic nerves contribute to pain [110, 111].
Traditional pain management begins with lifestyle changes. Cessation of alcohol abuse and smoking can prevent disease progression and provide pain relief [112, 113].
Analgesics are a mainstay of treatment. The WHO method can be used as a guide for pain relief starting with NSAIDs and progressing to strong opioids [114]. Tricyclic antidepressants such as amitriptyline and nortriptyline can be used with modest efficacy to reduce neuropathic pain [115]. Pregabalin has been shown to alleviate pain in CP [116, 117].
Pancreatic enzyme replacement therapy (PERT) can also be used to relieve pain, though the data remain controversial. Those with positive studies used uncoated pancreatic enzymes, which are not readily available [118, 119] and benefits may be related to placebo effect. A meta-analysis performed in 1997 showed no significant benefit of PERT to relieve pain [120]. However, PERT has relatively no side effects and is indicated in patients with exocrine pancreatic insufficiency (EPI) and steatorrhea [121].
Antioxidant therapy is another option for medical management of pain. Braganza et al. proposed that one of the mechanisms of CP is through increased oxidative stress leading to damage of pancreatic and acinar cells [122]. Current evidence suggests the decreased levels of antioxidants in patients with CP may be due to decrease intake and absorption secondary to pain and malabsorption, respectively [123]. A recent meta-analysis has shown reduction in pain symptoms with antioxidants consisting of organic selenium, ascorbic acid, beta-carotene, alpha-tocopherol, and methionine [123].
In addition, a recent study by Wu et al. [124] found that the use of simvastatin and atorvastatin were associated with an overall decrease risk in AP. Further subset analysis found a decrease in risk in patients with chronic alcohol abuse, suggesting the possibility of using simvastatin to prevent recurrent pancreatitis and subsequently, CP. A clinical trial is underway to test this possibility entitled “Simvastatin in reducing pancreatitis in patients with recurrent acute or CP” (ClinicalTrials.gov).
If medical therapy fails, more invasive measures of pain management can be utilized. Endoscopic decompression treatment with sphincterotomy and placement of stents can be performed in patients found to have obstructive stones or ductal stenosis [125, 126]. In 2002, Rosch et al. showed in their multicenter long-term study that two-thirds of patients were found post-procedure to experience long-term pain relief from 2 to 12 years [127]. In a recent meta-analysis, extracorporeal shock wave lithotripsy (ESWL) can also be used to relieve pain secondary to obstructive stones [128]. Additionally, EUS-guided celiac nerve block with steroids or alcohol can be used [129], though only ten percent of patients achieved pain relief for more than 24 weeks [130].
Surgical options exist when medical and minimally invasive therapies fail [131]. These procedures include decompression and drainage or resection.
Surgical decompression is reserved for patients with refractory pain with a dilated pancreatic duct. In 2011, Cahen et al. found that surgical decompression was found to have increased pain relief with 80% compared to 38% for endoscopic decompression [132], though the morbidity and mortality do increase with surgical treatments.
Resection is indicated in patients found to have pancreatic cancer or inflammatory mass causing post-obstructive CP, and in patients with small duct disease where a decompression procedure would not be helpful [112]. However, adverse effects including endocrine and exocrine insufficiency can occur. Novel therapies such as islet auto-transplantation have been developed to address endocrine insufficiency [112]. One study has shown success in preventing diabetes in ten of fourteen patients receiving more than 300,000 islets [133]. Exocrine insufficiency can be managed with PERT and vitamin supplementation.
Complications of Chronic Pancreatitis
CP can lead to other complications such as exocrine and endocrine insufficiencies. Exocrine insufficiency can occur with advanced disease, which clinically presents with weight loss, malabsorption, and steatorrhea. PERT and vitamin supplementation are the mainstays of treatment. Moreover, an adequate amount of calories should be taken each day to ensure weight gain [134]. If symptoms persist, median-chain triglycerides (MCT) can be used as they are directly absorbed by the intestinal mucosa even in the absence of lipase or bile salts [134].
Endocrine insufficiency can also occur secondary to islet cell destruction, which can ultimately lead to type 3c diabetes (T3cDM). T3cDM is generally managed in a similar fashion to type 2 diabetes with the initial use of metformin, but most patients with T3cDM will ultimately be insulin dependent [12, 113]. Additionally, type 3c diabetics are more prone to hypoglycemic episodes because glucagon secretion is altered [12]. Therefore, patients need to be educated on signs and symptoms of hypoglycemia and be prepared for management of acute hypoglycemic episodes.
Pseudocysts can form when fibrosis worsens. 39% of pseudocysts resolve spontaneously in patients with CP [135]. Pseudocysts, when large enough, can cause severe pain, infection, vascular compression, bleeding, or biliary stenosis. All of these findings are indications for endoscopic drainage. Surgery is indicated when endoscopic drainage fails or with large, multiple cysts [113].
Conclusion
This review has emphasized the current diagnostic and treatment modalities and the current diagnostic challenges we face with CP. An overall goal for the field should focus on the identification of disease pathogenesis and mechanisms of disease progression. The approach must be multidisciplinary and include the following types of information gathering:
-
1.
Investigate genetic and lifestyle factors associated with disease progression, especially those with “idiopathic” CP.
-
2.
Identify and validate novel biomarkers and imaging methods to facilitate early diagnosis and personalized treatment of recurring pancreatitis and CP with considerations into specific immune makers and enhanced measurements of fibrosis to personalize therapy for distinct subtype of patients.
-
3.
Develop novel, accurate and convenient tests to diagnose exocrine pancreatic insufficiency and monitor treatment response.
-
4.
Disseminate educational materials for use by medical practitioners to advise patients about life style changes needed to improve outcome.
-
5.
Determine the mechanism(s) of Type 3c diabetes and identify its distinction from other forms of diabetes.
-
6.
Determine the risk and prevalence of Type 3c diabetes along with its mechanism and optimal treatment strategies.
-
7.
Investigate new interventions for CP-related pain, including pharmacologic and non-pharmacologic therapies such as cognitive behavioral therapy.
-
8.
Develop therapeutics based on mechanisms of disease pathogenesis for the prevention and treatment of CP.
-
9.
Develop clinical trial methods and outcome measures for testing new therapeutics.
References
Yadav D, Timmons L, Benson JT, Dierkhising RA, Chari ST. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am J Gastroenterol. 2011;106:2192–2199.
Wang LW, Li ZS, Li SD, Jin ZD, Zou DW, Chen F. Prevalence and clinical features of chronic pancreatitis in China: a retrospective multicenter analysis over 10 years. Pancreas. 2009;38:248–254.
Mohan V, Farooq S, Deepa M. Prevalence of fibrocalculous pancreatic diabetes in Chennai in South India. JOP. 2008;9:489–492.
Balaji LN, Tandon RK, Tandon BN, et al. Prevalence and clinical features of chronic pancreatitis in southern India. Int J Pancreatol. 1994;15:29–34.
Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149:1731–1741.
Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358.
Lowenfels AB, Maisonneuve P, Grover H, et al. Racial factors and the risk of chronic pancreatitis. Am J Gastroenterol. 1999;94:790–794.
Kloppel G, Maillet B. The morphological basis for the evolution of acute pancreatitis into chronic pancreatitis. Virchows Arch A Pathol Anat Histopathol. 1992;420:1–4.
Mergener K, Baillie J. Chronic pancreatitis. Lancet. 1997;350:1379–1385.
Malka D, Hammel P, Sauvanet A, et al. Risk factors for diabetes mellitus in chronic pancreatitis. Gastroenterology. 2000;119:1324–1332.
Andersen DK, Andren-Sandberg A, Duell EJ, et al. Pancreatitis-diabetes-pancreatic cancer: summary of an NIDDK-NCI workshop. Pancreas. 2013;42:1227–1237. PMCID:3878448.
Gudipaty L, Rickels MR. Pancreatogenic (Type 3c) Diabetes. 2015.
Ammann RW, Akovbiantz A, Largiader F, Schueler G. Course and outcome of chronic pancreatitis: longitudinal study of a mixed medical-surgical series of 245 patients. Gastroenterology. 1984;86:820–828.
Lankisch PG, Lohr-Happe A, Otto J, Creutzfeldt W. Natural course in chronic pancreatitis: pain, exocrine and endocrine pancreatic insufficiency and prognosis of the disease. Digestion. 1993;54:148–155.
Comfort HW, Gambill EE, Baggenstoss AH. Chronic relapsing pancreatitis: a study of 29 cases without associated disease of the biliary or gastrointestinal tract. Gastroenterology. 1946;6:239–285.
Singer MV, Gyr K, Sarles H. Revised classifi cation of pancreatitis. Report of the Second International Symposium on the classification of pancreatitis in Marseille, France, March 28–30, 1984. Gastroenterology. 1985;89:683–685.
Lankisch PG, Breuer N, Bruns A, et al. Natural history of acute pancreatitis: a long-term population based study. Am J Gastroenterol. 2009;104:2797–2805. quiz 2806.
Nojgaard C, Becker U, Matzen P, Andersen JR, Holst C, Bendtsen F. Progression from acute to chronic pancreatitis: prognostic factors, mortality, and natural course. Pancreas. 2011;40:1195–1200.
Yadav D, O’Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. Am J Gastroenterol. 2012;107:1096–1103.
Comfort MW, Steinberg AG. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology. 1952;21:54–63.
Stevens T, Conwell DL, Zuccaro G. Pathogenesis of chronic pancreatitis: an evidence-based review of past theories and recent developments. Am J Gastroenterol. 2004;99:2256–2270.
Kloppel G, Maillet B. Chronic pancreatitis: evolution of the disease. Hepatogastroenterol. 1991;38:408–412.
Schneider A, Whitcomb DC. Hereditary pancreatitis: a model for inflammatory diseases of the pancreas. Best Pract Res Clin Gastroenterol. 2002;16:347–363.
Takeyama Y. Long-term prognosis of acute pancreatitis in Japan. Clin Gastroenterol Hepatol. 2009;7:S15–S17.
Whitcomb DC, Preston RA, Aston CE, et al. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology. 1996;110:1975–1980.
Irving HM, Samokhvalov AV, Rehm J. Alcohol as a risk factor for pancreatitis. A systematic review and meta-analysis. JOP. 2009;10:387–392.
Samokhvalov AV, Rehm J, Roerecke M. Alcohol consumption as a risk factor for acute and chronic pancreatitis: a systematic review and a series of meta-analyses. EBioMedicine. 2015;2:1996–2002.
Corrao G, Bagnardi V, Zambon A, Arico S. Exploring the dose-response relationship between alcohol consumption and the risk of several alcohol-related conditions: a meta-analysis. Addiction. 1999;94:1551–1573.
Setiawan VW, Pandol SJ, Porcel J, et al. Prospective study of alcohol drinking, smoking, and pancreatitis: the multiethnic cohort. Pancreas. 2016;45:819–825.
Pandol SJ, Periskic S, Gukovsky I, et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology. 1999;117:706–716.
Yuan J, Lugea A, Zheng L, et al. Protein kinase D1 mediates NF-kappaB activation induced by cholecystokinin and cholinergic signaling in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1190–G1201.
Pandol SJ, Gorelick FS, Lugea A. Environmental and genetic stressors and the unfolded protein response in exocrine pancreatic function—a hypothesis. Front Physiol. 2011;2:8.
Lugea A, Tischler D, Nguyen J, et al. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology. 2011;140:987–997.
Lugea A, Waldron RT, Pandol SJ. Pancreatic adaptive responses in alcohol abuse: role of the unfolded protein response. Pancreatology. 2015;15:S1–S5.
Yadav D, Eigenbrodt ML, Briggs MJ, Williams DK, Wiseman EJ. Pancreatitis: prevalence and risk factors among male veterans in a detoxification program. Pancreas. 2007;34:390–398. [PubMed:17446836].
Whitcomb DC, Larusch J, Krasinskas AM, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet. 2012;44:1349–1354.
Van den Bossche J, Laoui D, Morias Y, et al. Claudin-1, claudin-2 and claudin-11 genes differentially associate with distinct types of anti-inflammatory macrophages in vitro and with parasite- and tumour-elicited macrophages in vivo. Scand J Immunol. 2012;75:588–598.
Andriulli A, Botteri E, Almasio PL, Vantini I, Uomo G, Maisonneuve P. For the ad hoc committee of the Italian Association for the study of the pancreas. Smoking as a cofactor for causation of chronic pancreatitis: a meta-analysis. Pancreas. 2010;39:1205–1210.
Talamini G, Falconi M, Bassi C, et al. Incidence of cancer in the course of chronic pancreatitis. Am J Gastroenterol. 1999;94:1253–1260.
Howes N, Lerch MM, Greenhalf W, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2:252–261.
Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141–145.
Perrault J. Hereditary pancreatitis. Gastroenterol Clin North Am. 1994;23:743–752.
Schneider A, Barmada MM, Slivka A, Martin JA, Whitcomb DC. Clinical characterization of patients with idiopathic chronic pancreatitis and SPINK1 Mutations. Scand J Gastroenterol. 2004;39:903–904.
Rosendahl J, Witt H, Szmola R, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82.
Hegyi P, Pandol S, Venglovecz V, Rakonczay Z. The acinar-ductal tango in the pathogenesis of acute pancreatitis. Gut. 2011;60:544–552.
Sharer N, Schwarz M, Malone G, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–652.
Maléth J, Balázs A, Pallagi P, et al. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology. 2015;148:427–439.
Warshaw AL, Richter JM, Schapiro RH. The cause and treatment of pancreatitis associated with pancreas divisum. Ann Surg. 1983;198:443–452.
Bertin C, Pelletier AL, Vullierme MP, et al. Pancreas divisum is not a cause of pancreatitis by itself but acts as a partner of genetic mutations. Am J Gastroenterol. 2012;107:311–317.
Layer P, Yamamoto H, Kalthoff L, et al. The different courses of early and late onset idiopathic and alcoholic chronic pancreatitis. Gastroenterology. 1994;107:1481–1487.
Ammann RW. Alcohol and non-alcohol induced pancreatitis: clinical aspects (chapter 16). In: Burns GP, Bank S, eds. Disorders of the pancreas: current issues in diagnosis and management. Philadelphia: McGraw Hill; 1992:253–271.
Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol. 1998;275:G1402–G1414.
Blinman TA, Gukovsky I, Mouria M, et al. Activation of pancreatic acinar cells on isolation from tissue: cytokine upregulation via p38 MAP kinase. Am J Physiol Cell Physiol. 2000;279:C1993–C2003.
Zaninovic V, Gukovskaya AS, Gukovsky I, Mouria M, Pandol SJ. Cerulein upregulates ICAM-1 in pancreatic acinar cells, which mediates neutrophil adhesion to these cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G666–G676.
Habtezion A. Inflammation in acute and chronic pancreatitis. Curr Opin Gastroenterol. 2015;31:395–399.
Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1199–1209.
Apte MV, Haber PS, Darby SJ, et al. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534–541.
Vonlaufen A, Phillips PA, Xu Z, et al. Withdrawal of alcohol promotes regression while continued alcohol intake promotes persistence of LPS-induced pancreatic injury in alcohol-fed rats. Gut. 2011;60:238–246.
Vonlaufen A, Xu Z, Daniel B, et al. Bacterial endotoxin: a trigger factor for alcoholic pancreatitis? Evidence from a novel, physiologically relevant animal model. Gastroenterology. 2007;133:1293–1303.
Apte MV, Phillips PA, Fahmy RG, et al. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology. 2000;118:780–794.
Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–59.
Apte MV, Pirola RC, Wilson JS. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol. 2012;3:344.
Van Laetham JL, Deviere J, Resibois A, et al. Localization of transforming growth factor beta 1 and its latent binding protein in human chronic pancreatitis. Gastroenterology. 1995;108:1873–1881.
Vogelmann R, Ruf D, Wagner M, et al. Effects of fibrogenic mediators on the development of pancreatic fibrosis in a TGF-β1 transgenic mouse model. Am J Phys. 2001;280:G164–G172.
Xue J, Sharma V, Hsieh MH, et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. 2015;6:7158.
Xue J, Zhao Q, Sharma V, et al. Aryl hydrocarbon receptor ligands in cigarette smoke induce production of interleukin-22 to promote pancreatic fibrosis in models of chronic pancreatitis. Gastroenterology. 2016;151:1206–1217.
Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med.. 1995;332:1482–1490.
Manjari KS, Jyothy A, Vidyasagar A, Prabhakar B, Nallari P, Venkateshwari A. Matrix metalloproteinase-9, transforming growth factor-β1, and tumor necrosis factor-α plasma levels in chronic pancreatitis. Indian J Gastroenterol. 2013;32:103–107.
Yasuda M, Ito T, Oono T, et al. Fractalkine and TGF-β1 levels reflect the severity of chronic pancreatitis in humans. World J Gastroenterol. 2008;14:6488–6495.
Alpern MB, Sandler MA, Kellman GM, et al. Chronic pancreatitis: ultrasonic features. Radiology. 1985;155:215–219.
Luetmer PH, Stephens DH, Ward EM. Chronic pancreatitis: reassessment with current CT. Radiology. 1989;171:353–357.
Conwell DL, Lee LS, Yadav D. American Pancreatic Association Practice Guidelines in Chronic Pancreatitis: evidence-based report on diagnostic guidelines. Pancreas. 2014;43:1143–1162.
Balci NC, Alkaade S, Magas L, Momtahen AJ, Burton FR. Suspected chronic pancreatitis with normal MRCP: findings on MRI in correlation with secretin MRCP. J Magn Reson Imaging JMRI. 2008;27:125–131.
Tirkes T, Fogel EL, Sherman S, et al. Detection of exocrine dysfunction by MRI in patients with early chronic pancreatitis. Abdom Radiol. 2017;42:544–551.
Stevens T. Role of endoscopic ultrasonography in the diagnosis of acute and chronic pancreatitis. Gastrointest Endosc Clin N Am. 2013;23:735–747.
Morris-stiff G, Webster P, Frost B, Lewis WG, Puntis MC, Roberts SA. Endoscopic ultrasound reliably identifies chronic pancreatitis when other imaging modalities have been non-diagnostic. JOP. 2009;10:280–283.
Wiersema MJ, Wiersema LM. Endosonography of the pancreas: normal variation versus changes of early chronic pancreatitis. Gastrointest Endosc Clin N Am. 1995;5:487–496.
Chong AK, Hawes RH, Hoffman BJ, Adams DB, Lewin DN, Romagnuolo J. Diagnostic performance of EUS for chronic pancreatitis: a comparison with histopathology. Gastrointest Endosc. 2007;65:808–814.
Albashir S, Bronner MP, Parsi MA, Walsh RM, Stevens T. Endoscopic ultrasound, secretin endoscopic pancreatic function test, and histology: correlation in chronic pancreatitis. Am J Gastroenterol. 2010;105:2498–2503.
Iglesias-garcia J, Domínguez-muñoz JE, Castiñeira-alvariño M, Luaces-regueira M, Lariño-noia J. Quantitative elastography associated with endoscopic ultrasound for the diagnosis of chronic pancreatitis. Endoscopy. 2013;45:781–788.
Itoh Y, Itoh A, Kawashima H, et al. Quantitative analysis of diagnosing pancreatic fibrosis using EUS-elastography (comparison with surgical specimens). J Gastroenterol. 2014;49:1183–1192.
Janssen J, Schlörer E, Greiner L. EUS elastography of the pancreas: feasibility and pattern description of the normal pancreas, chronic pancreatitis, and focal pancreatic lesions. Gastrointest Endosc. 2007;65:971–978.
Wallace MB, Hawes RH, Durkalski V, et al. The reliability of EUS for the diagnosis of chronic pancreatitis: interobserver agreement among experienced endosonographers. Gastrointest Endosc. 2001;53:294–299.
Adler DG, Lichtenstein D, Baron TH, et al. The role of endoscopy in patients with chronic pancreatitis. Gastrointest Endosc. 2006;63:933–937.
Axon AT, Classen M, Cotton PB, et al. Pancreatography in chronic pancreatitis: international definitions. Gut. 1984;25:1107–1112.
Vitale GC, Davis BR, Zavaleta C, Vitale M, Fullerton JK. Endoscopic retrograde cholangiopancreatography and histopathology correlation for chronic pancreatitis. Am Surg. 2009;75:649–653.
Varadarajulu S, Eltoum I, Tamhane A, Eloubeidi MA. Histopathologic correlates of noncalcific chronic pancreatitis by EUS: a prospective tissue characterization study. Gastrointest Endosc. 2007;66:501–509.
LeBlanc JK, Chen JH, Al-Haddad M, et al. Endoscopic ultrasound and histology in chronic pancreatitis: how are they associated? Pancreas. 2014;43:440–444.
Bhutani MS, Arantes VN, Verma D, et al. Histopathologic correlation of endoscopic ultrasound findings of chronic pancreatitis in human autopsies. Pancreas. 2009;38:820–824.
Trikudanathan G, Vega-Peralta J, Malli A, et al. Diagnostic performance of endoscopic ultrasound (EUS) for non-calcific chronic pancreatitis (NCCP) based on histopathology. Am J Gastroenterol. 2016;111:568–574.
Rajan E, Clain JE, Levy MJ, et al. Age-related changes in the pancreas identified by EUS: a prospective evaluation. Gastrointest Endosc. 2005;61:401–406.
Yusoff IF, Sahai AV. A prospective, quantitative assessment of the effect of ethanol and other variables on the endosonographic appearance of the pancreas. Clin Gastroenterol Hepatol. 2004;2:405–409.
Chantarojanasiri T, Hirooka Y, Ratanachu-Ek T, Kawashima H, Ohno E, Goto H. Evolution of pancreas in aging: degenerative variation or early changes of disease? J Med Ultrason. 2001;2015:177–183.
Hastier P, Buckley MJ, Francois E, et al. A prospective study of pancreatic disease in patients with alcoholic cirrhosis: comparative diagnostic value of ERCP and EUS and long-term significance of isolated parenchymal abnormalities. Gastrointest Endosc. 1999;49:705–709.
Chowdhury RS, Forsmark CE. Review article: pancreatic function testing. Aliment Pharmacol Ther. 2003;17:733–750.
DiMagno EP, Go VL, Summerskill WH. Relations between pancreatic enzyme ouputs and malabsorption in severe pancreatic insufficiency. N Engl J Med. 1973;288:813–815.
Bo-linn GW, Fordtran JS. Fecal fat concentration in patients with steatorrhea. Gastroenterology. 1984;87:319–322.
Gullo L, Ventrucci M, Tomassetti P, Migliori M, Pezzilli R. Fecal elastase 1 determination in chronic pancreatitis. Dig Dis Sci. 1999;44:210–213.
Walkowiak J, Herzig KH, Strzykala K, Przyslawski J, Krawczynski M. Fecal elastase-1 is superior to fecal chymotrypsin in the assessment of pancreatic involvement in cystic fibrosis. Pediatrics. 2002;110:e7.
Amann ST, Bishop M, Curington C, Toskes PP. Fecal pancreatic elastase 1 is inaccurate in the diagnosis of chronic pancreatitis. Pancreas. 1996;13:226–230.
Jacobson DG, Curington C, Connery K, Toskes PP. Trypsin-like immunoreactivity as a test for pancreatic insufficiency. N Engl J Med. 1984;310:1307–1309.
Laterza L, Scaldaferri F, Bruno G, et al. Pancreatic function assessment. Eur Rev Med Pharmacol Sci. 2013;17:65–71.
Braden B. (13) C breath tests for the assessment of exocrine pancreatic function. Pancreas. 2010;39:955–959.
van Dijk-van Aalst K, Van Den Driessche M, van Der Schoor S, et al. 13C mixed triglyceride breath test: a noninvasive method to assess lipase activity in children. J Pediatr Gastroenterol Nutr. 2001;32:579–585.
Loser C, Brauer C, Aygen S, Hennemann O, Folsch UR. Comparative clinical evaluation of the 13C mixed triglyceride breath test as an indirect pancreatic function test. Scand J Gastroenterol. 1998;33:327–334.
Gyr K, Agrawal NM, Felsenfeld O, Font RG. Comparative study of secretin and Lundh tests. Am J Dig Dis. 1975;20:506–512.
Lieb JG II, Draganov PV. Pancreatic function testing: here to stay for the 21st century. World J Gastroenterol. 2008;14:3149–3158.
Ketwaroo G, Brown A, Young B, et al. Defining the accuracy of secretin pancreatic function testing in patients with suspected early chronic pancreatitis. Am J Gastroenterol. 2013;108:1360–1366.
Hart PA, Topazian M, Raimondo M, et al. Endoscopic pancreas fluid collection: methods and relevance for clinical care and translational science. Am J Gastroenterol. 2016;111:1258–1266.
Demir IE, Friess H, Ceyhan GO. Neural plasticity in pancreatitis and pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2015;12:649–659.
Uc A, Andersen DK, Bellin MD, et al. Chronic pancreatitis in the 21st century—research challenges and opportunities: summary of a National Institute of Diabetes and Digestive and Kidney Diseases Workshop. Pancreas. 2016;45:1365–1375.
Goulden MR. The pain of chronic pancreatitis: a persistent clinical challenge. Br J Pain. 2013;7:8–22.
De-madaria E, Abad-gonzález A, Aparicio JR, et al. The Spanish Pancreatic Club’s recommendations for the diagnosis and treatment of chronic pancreatitis: part 2 (treatment). Pancreatology. 2013;13:18–28.
Ventafridda V, Tamburini M, Caraceni A, De conno F, Naldi F. A validation study of the WHO method for cancer pain relief. Cancer. 1987;59:850–856.
Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet. 2009;374:1252–1261.
Olesen SS, Bouwense SA, Wilder-smith OH, Van goor H, Drewes AM. Pregabalin reduces pain in patients with chronic pancreatitis in a randomized, controlled trial. Gastroenterology. 2011;141:536–543.
Bouwense SA, Olesen SS, Drewes AM, et al. Effects of pregabalin on central sensitization in patients with chronic pancreatitis in a randomized, controlled trial. PLoS One. 2012;7:e42096.
Slaff J, Jacobson D, Tillman CR, Curington C, Toskes P. Protease-specific suppression of pancreatic exocrine secretion. Gastroenterology. 1984;87:44–52.
Isaksson G, Ihse I. Pain reduction by an oral pancreatic enzyme preparation in chronic pancreatitis. Dig Dis Sci. 1983;28:97e102.
Brown A, Hughes M, Tenner S, Banks PA. Does pancreatic enzyme supplementation reduce pain in patients with chronic pancreatitis: a meta-analysis. Am J Gastroenterol. 1997;92:2032–2035.
Taylor JR, Gardner TB, Waljee AK, DiMagno MJ, Schoenfeld PS. Systematic review: efficacy and safety of pancreatic enzyme supplements for exocrine pancreatic insufficiency. Aliment Pharmacol Ther. 2010;31:57–72.
Braganza JM. Pancreatic disease: a casualty of hepatic ‘detoxification’? Lancet. 1983;2:1000–1002.
Ahmed Ali U, Jens S, Busch OR, et al. Antioxidants for pain in chronic pancreatitis. Cochrane Database Syst Rev. 2014;(8):CD008945.
Wu BU, Pandol SJ, Liu IL. Simvastatin is associated with reduced risk of acute pancreatitis: findings from a regional integrated healthcare system. Gut. 2015;64:133–138.
Gabbrielli A, Pandolfi M, Mutignani M, et al. Efficacy of main pancreatic-duct endoscopic drainage in patients with chronic pancreatitis, continuous pain, and dilated duct. Gastrointest Endosc. 2005;61:576–581.
Dumonceau JM, Deviere J, Le MO, et al. Endoscopic pancreatic drainage in chronic pancreatitis associated with ductal stones: long-term results. Gastrointest Endosc. 1996;43:547–555.
Rösch T, Daniel S, Scholz M, et al. Endoscopic treatment of chronic pancreatitis: a multicenter study of 1000 patients with long-term follow-up. Endoscopy. 2002;34:765–771.
Guda NM, Partington S, Freeman ML. Extracorporeal shock wave lithotripsy in the management of chronic calcific pancreatitis: a meta-analysis. JOP. 2005;6:6–12.
Michaels AJ, Draganov PV. Endoscopic ultrasonography guided celiac plexus neurolysis and celiac plexus block in the management of pain due to pancreatic cancer and chronic pancreatitis. World J Gastroenterol. 2007;13:3575–3580.
Santosh D, Lakhtakia S, Gupta R, et al. Clinical trial: a randomized trial comparing fluoroscopy guided percutaneous technique vs. endoscopic ultrasound guided technique of coeliac plexus block for treatment of pain in chronic pancreatitis. Aliment Pharmacol Ther. 2009;29:979–984.
Bellin MD, Freeman ML, Gelrud A, et al. Total pancreatectomy and islet auto transplantation in chronic pancreatitis: recommendations from PancreasFest. Pancreatology. 2014;14:27–35.
Cahen DL, Gouma DJ, Laramée P, et al. Long-term outcomes of endoscopic vs surgical drainage of the pancreatic duct in patients with chronic pancreatitis. Gastroenterology. 2011;141:1690–1695.
Wahoff DC, Papalois BE, Najarian JS, et al. Autologous islet transplantation to prevent diabetes after pancreatic resection. Ann Surg. 1995;222:562–575.
Rasmussen HH, Irtun O, Olesen SS, Drewes AM, Holst M. Nutrition in chronic pancreatitis. World J Gastroenterol. 2013;19:7267–7275.
Talar-Wojnarowska R, Wozniak B, Pazurek M, Malecka-Panas E. Outcome of pseudocysts complicating chronic pancreatitis. Hepatogastroenterology. 2010;57:631–634.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
This work was supported by the following US federal grants: CA163200, AA11999, DK098108, DoD PR140717P2, DK108314, DCP NWU2014-04-01, W81XWH-16-PRMRP-DA. Dr. Pandol is a founder of Avenzoar Pharmaceuticals and a consultant for Allergan and SunBiopharma. There is no information in this work representing a commercial conflict of interest. Dr. Lew and Dr. Afghani do not have any conflict of interests.
Rights and permissions
About this article

Cite this article
Lew, D., Afghani, E. & Pandol, S. Chronic Pancreatitis: Current Status and Challenges for Prevention and Treatment. Dig Dis Sci 62, 1702–1712 (2017). https://doi.org/10.1007/s10620-017-4602-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-017-4602-2