
Abstract
Purpose
Plecanatide, an analogue of uroguanylin, activates the guanylate cyclase C (GC-C) receptor found on the GI mucosal epithelial cells, leading to secretion of fluid, facilitating bowel movements. Plecanatide is being investigated as a potential treatment for constipating GI disorders. The aim of this investigation was to assess the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of single doses of plecanatide in healthy volunteers.
Methods
A total of 72 healthy volunteers at a single site were randomized in 9 cohorts to receive oral plecanatide or placebo from 0.1 to 48.6 mg. Plasma PK samples were collected pre-dose and post-dose. PD assessments included time to first stool, stool frequency, and stool consistency using the Bristol Stool Form Scale. All adverse events were documented.
Results
Plecanatide was safe and well-tolerated at all dose levels. A total of 17 of 71 subjects (23.9 %) reported 25 treatment-emergent adverse events (TEAEs) during the study. The number of TEAEs reported by subjects who received plecanatide or placebo was comparable (24.5 vs. 22.2 %, respectively). There were no dose-related increases in TEAEs or any SAEs reported. No measurable systemic absorption of oral plecanatide was observed at any of the oral doses studied, utilizing an assay sensitive down to 1 ng/mL.
Conclusions
Plecanatide, an oral GC-C agonist, acting locally within the GI tract without measurable systemic exposure, was safe and well-tolerated in single doses up to 48.6 mg. The study was not powered for statistical analyses, but trends in PD parameters supported continued clinical development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic functional constipation (CC) and irritable bowel syndrome-constipation predominant (IBS-C) are the most common gastrointestinal (GI) disorders, accounting for up to 19 % of total visits to primary care providers [1, 2]. These GI disorders are characterized by abdominal discomfort, cramping and changes in bowel function—including bloating, gas, and constipation [3]. Abdominal pain due to the hypersensitivity of the colon to mechanical stimuli is also a major clinical symptom for IBS-C. There are currently two FDA-approved drugs to treat CC and IBS-C in the US: lubiprostone (Amitiza®), an activator of the chloride type 2 ion channel [4], and linaclotide (Linzess®), a guanylate cyclase C (GC-C) agonist, which was recently approved to treat both CC and IBS-C [5]. There continues to be a strong need for additional effective and well-tolerated therapies, as patients with CC and IBC-C are generally not completely satisfied with available treatments [6].
Agonists of GC-C are a new class of drugs to treat GI disorders [7, 8]. The physiological agonists of GC-C receptors are the natriuretic peptides, uroguanylin and guanylin, which are structurally similar to the heat-stable enterotoxin (ST peptide) produced and secreted by the pathogenic Escherichia coli (E. coli) responsible for traveler’s diarrhea [9, 10]. Binding of these agonists to GC-C receptors stimulates intracellular production of cyclic guanosine monophosphate (cGMP), leading to activation of the cystic fibrosis transmembrane conductance regulator (CFTR), the apical ion channel responsible for efflux of chloride ions from enterocytes lining the GI tract, resulting in a net efflux of water into the intestinal lumen [11, 12]. Uroguanylin activates GC-C receptors in a pH-dependent fashion, regulated by mucosal acidity in the proximal intestine, producing a sufficient volume of water to normalize bowel movements. In contrast, ST peptide, [13] is designed to activate GC-C in a pH-independent manner giving rise to excessive fluid secretion and diarrhea. The key difference between uroguanylin and that of ST peptide of E. coli is the presence of two charged aspartate amino acids within the N terminus of uroguanylin which regulate binding affinity of uroguanylin to GC-C in a pH-dependent manner [13]. The structure of plecanatide is virtually identical to that of uroguanylin except for the replacement of the penultimate aspartate on N-terminus with a glutamate amino acid. Consequently, plecanatide demonstrates the same pH dependency to its binding to the GC-C receptor as is observed with uroguanylin.
Orally administered plecanatide is expected to act in an identical fashion to uroguanylin, binding and activating GC-C receptors within the GI tract, leading to activation of the CFTR. The resultant efflux of chloride ions is expected to lead to secretion of fluid into the intestinal lumen, facilitating bowel movements. Plecanatide has the potential to improve on current therapies to treat CC and IBS-C.
The purpose of this first-in-human clinical study was to characterize the safety and tolerability of single ascending doses of plecanatide in adult healthy volunteers. Additionally, pharmacokinetics (PK) and pharmacodynamics (PD) parameters were assessed.
Methods
Study Design, Randomization, and Eligibility Criteria
This study was a Phase 1, first-in-human, single-site, randomized, double-blind, placebo-controlled, single ascending-dose study evaluating the effects of orally administered plecanatide in healthy volunteers. Subjects who met all criteria for enrollment were sequentially assigned to dosing cohorts. Within a cohort, each subject was given a unique number that assigned him/her to receive treatment (plecanatide or placebo, at a 3:1 ratio) according to the pre-specified randomization schedule generated by a biostatistician who provided the schedule to the unblinded pharmacist. Participants were enrolled at a single site (Comprehensive Phase One, Miramar, Florida, between June 2, 2008 and August 17, 2008). The study was approved by an Independent Investigational Review Board (Plantation, FL, USA). This study was conducted in accordance with Good Clinical Practices and the International Conference on Harmonization (ICH) guidelines, and the Declaration of Helsinki.
Healthy male and post-menopausal female volunteers between the ages of 18 and 64 years with a body mass index (BMI) between 18 and 29 kg/m2 were recruited for the study. All participants signed the informed consent form. Subjects abstained from caffeinated beverages, alcohol, and nicotine for a period of 36 h prior to dosing through 48 h post-dose, and abstained from and had no clinically-indicated requirement for supplemental fiber, laxatives, or herbal or dietary supplements intended to treat constipation within 30 days of study entry.
Following the completion of each dose level, blinded safety and tolerability data through 48 h post-dose (e.g., laboratory results, AEs, ECGs, vital sign measurement, etc.) were reviewed by the Investigator and sponsor. Decisions regarding dose escalation or dose adjustment were made after reviewing safety and tolerability data for each cohort, and were documented. As defined by NCI-CTCAE, a maximum tolerated dose is defined as the next highest dose to the one at which three or more subjects experience a study drug-related Grade 3 dose-limiting toxicity, or at least one subject experiences a Grade 4 dose-limiting toxicity.
Study Drug Administration
For each cohort of 8 subjects (n = 6 plecanatide and n = 2 placebo), a concentrated stock solution of plecanatide and a separate phosphate-buffered saline (PBS) solution were used to prepare placebo and plecanatide doses by an unblinded registered pharmacist at the investigational site. The concentrated stock solution was prepared by adding plecanatide drug substance to an appropriate volume of PBS. Plecanatide is an amorphous, white colored and tasteless peptide. The peptide may be dosed as a solution in water without affecting its palatability. However, the room temperature stability of plecanatide solution is superior in PBS as compared to that in water. Thus, plecanatide solution in PBS was used in this study. The administration solution for each subject receiving plecanatide was prepared from this concentrated stock solution by adding an appropriate volume of stock solution to additional PBS to give a total volume of 240 mL. Placebo doses for administration were prepared using solely 240 mL of PBS solution without active drug. The concentrated stock solutions were prepared and refrigerated approximately 1 h prior to preparation of the administration solutions, which occurred approximately 1 h prior to administration of the study drug. Immediately following administration of the 240 mL dose of the study drug, subjects consumed 60 mL of water from the same dosing cup.
Subjects had been fasting for at least 10 h prior to the time they received the study drug. A total of 180 mL water were given 2 h after study drug administration. A standardized meal was provided 4 h after study drug administration. Water was allowed ad libitum after consumption of a meal on the day of dosing through to discharge.
PK Sampling and Quantification of Plecanatide
Blood samples for determination of plecanatide concentrations in plasma were obtained at Time 0 (pre-dose) and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h post-dosing, respectively, on Days 1 through 3. Plasma concentrations of plecanatide were determined using analysis methods validated by Pyxant Labs (Colorado Springs, CO, USA) with a lower limit of quantification (LLOQ) of 10 mg/mL. Following the performance of this study, blood samples from the 48.6 mg dose level were also re-analyzed with an improved method with a LLOQ of 1 ng/mL.
Daily Stool Diaries
Subjects recorded details regarding their bowel movements for 7 consecutive days during the 14-day screening period. Details included date and time of each bowel movement and the description of the stool in each bowel movement using the Bristol Stool Form Scale [BSFS] [14]. This scale rates the consistency of stools from 1 to 7, with higher numbers indicating looser stools. After dosing, subjects recorded the date and time of each bowel movement and stool consistency (BSFS score) within the 48-h period post-dose.
Statistical Analysis
Pharmacodynamics assessments included time to first bowel movement, stool consistency (over the 48-h period) using the BSFS Scale, and stool frequency (over the 48-h period). Time to first bowel movement was assessed for each subject (in hours) and the mean time was calculated for subjects receiving plecanatide in each cohort. Post hoc analyses assessed stool consistency (mean BSFS score) and stool frequency [mean # bowel movements (BMs)/day] for each subject, averaging values at pre-dose (at least 7 days prior to treatment) and post-dose from 0 to 24 h and from 24 to 48 h. Mean stool consistency for each cohort was based on each subject’s mean BSFS score for BMs reported within each timeframe (pre-dose, 0 to 24 h post-dose, 24 to 48 h post-dose, and first BM within 48 h post-dose). Mean stool frequency for each cohort was based on each subject’s mean number of bowel movements per day within each timeframe (at least 7 days pre-dose, and post-dose from 0 to 24 h and 24 to 48 h). Means + SEM are reported.
The sample size chosen for the study was based upon precedent set by other PK studies of a similar nature, and was not based on power calculations for any pharmacodynamic or safety parameter.
The verbatim AE terms were coded using Medical Dictionary for Regulatory Activities, version 10.1, and classified by system organ class and preferred term. All TEAE data recorded on the electronic case report form were tabulated and analyzed.
Results
Participants, Study Conduct, and Completion Rate
This is a phase I study with limited numbers of adult healthy volunteers, and it was not powered to statistically assess potential pharmacodynamic (PD) activities of plecanatide. A total of 72 adult healthy volunteers were screened and randomized to receive plecanatide (n = 54) or placebo (n = 18). The study drug was administered as a single dose to 8 subjects (n = 6 plecanatide and n = 2 placebo) at each of the 9 dose levels, including 0.1, 0.3, 0.9, 2.7, 5.4, 8.1, 16.2, 24.3, and 48.6 mg plecanatide, respectively. At the 16.2 mg dose level, one subject was randomized to receive plecanatide, but did not meet inclusion/exclusion criteria due to a positive test for occult blood, and was consequently not dosed. As a result, a total of n = 53 subjects were dosed with plecanatide in this study (Fig. 1).

Study flow diagram. Seventy-two healthy volunteers were screened and sequentially assigned to 9 single-ascending dose cohorts. Within each cohort, subjects were randomized 3:1 to receive plecanatide or placebo. One subject failed screening and was not dosed. All remaining 71 subjects completed the study procedures and are included in the analysis
All 71 dosed subjects completed the study between June 2, 2008 and August 17, 2008. No further dosing of volunteers occurred after completion of the 48.6 mg cohort, although a Maximum Tolerated Dose (MTD) was not declared. Demographic data of subjects in each cohort and of the total population are found in Table 1. The demographic data show that all treatment groups and the placebo group included subjects with similar ages, heights, weights, and body mass indices.
Plasma Concentration of Plecanatide
All 53 subjects who received a single dose of plecanatide were evaluated for plasma concentrations of plecanatide using blood samples collected at Time = 0 (pre-dose), and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h post-dosing, respectively, on Days 1 through 3. Notably, no plecanatide was detected in any plasma sample collected during the study using a validated assay with a lower limit of quantitation (LLOQ) of 10 ng/mL. Subsequently, a more sensitive bioanalytical method with LLOQ sensitivity down to 1 ng/mL for plecanatide was developed and used to reanalyze samples collected from subjects at the highest plecanatide dose (48.6 mg). Again, no plecanatide was detected in any of these samples.
Time to First Bowel Movement
The mean time to first bowel movement was highly variable across the plecanatide dose range generally occurring within 24 h at all doses except for the 5.1 mg cohort. With the exception of the 2.7 mg plecanatide dose, higher doses of plecanatide trended to lower mean times to first bowel movement more than lower doses of plecanatide, with the lowest mean time post-dose observed at the 16.2 mg plecanatide dose level (6.3 h) followed by the 24.3 mg plecanatide dose level with a mean time to first bowel movement of 7 h.
Stool Consistency (Bristol Stool Form Scale Scores)
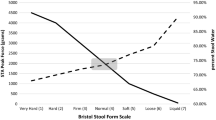
As shown in Fig. 2a, mean pre-dose BSFS scores were comparable across all the dosing groups, and were representative of a “normal” stool consistency, with values between 3 and 5 on a 7-point scale. Mean BSFS scores were also analyzed for subjects reporting post-dose BMs between 0 and 24 h, and between 24 and 48 h, respectively (Fig. 2a). Because not all subjects had BMs to report within each timeframe, the number of subjects contributing to each mean varied between 3 and 6. In general, the mean BSFS scores between 0 and 24 h were higher compared to pre-dose as well as to the 24–48 h time period for each plecanatide dose level ≥0.9 mg.

Stool consistency using Bristol Stool Form Scale. Subjects scored the consistency of each BM prior to dosing and for 48 h after dosing using the 7-point Bristol Stool Form Scale, which rates stools from 1 (hard lumps) to 7 (watery). a The mean BSFS (+SEM) is shown for subjects reporting BMs at pre-dose, 0–24 h post-dose, and 24–48 h post-dose. b The mean BSFS (+SEM) is shown for the first BM reported post-dose. In both graphs, the dashed lines delineate the range of BSFS considered normal which is generally between scores of 3 and 5
The mean BSFS scores of the first bowel movement post-dose (Fig. 2b) demonstrated a trend towards higher scores with increasing plecanatide dose, although scores remained within the normal range for the majority of doses.
Stool Frequency
The mean pre-dose frequency of bowel movements per day was similar across the plecanatide dose range and comparable to subjects receiving placebo. However, post-dose mean frequencies were highly variable across the plecanatide dose range with no trends observed between the time periods. Comparisons of each cohort’s mean frequency from 0 to 24 h post-dose do not reveal any dose-related trends. The mean change in stool frequency was not significantly different amongst the placebo and plecanatide dose levels post-dose within any of the observed time periods.
Adverse Events
A total of 17 (23.9 % of 71) subjects reported 25 treatment-emergent adverse events (TEAEs) during the study. These TEAEs, outlined in Table 2, were reported in almost equal percentages by subjects receiving plecanatide (24.5 % of 53) or placebo (22.2 % of 18). No serious adverse events were observed.
Diarrhea was the most prevalent TEAE reported by subjects. Diarrhea was defined in the protocol by the NCI-CTCAE v.3.0 as an increase in the number of bowel movements over baseline in a 24-h period. Diarrhea was reported in a slightly lower percentage of subjects receiving plecanatide (8 of 53, 15.1 %) as compared to the percentage in subjects receiving placebo (3 of 18, 16.7 %). The incidence of diarrhea in patients receiving plecanatide did not appear to be dose-related as no clear trend of increasing diarrhea was observed at higher plecanatide doses. In fact, of the eight volunteers on plecanatide who reported diarrhea, seven of the eight incidents of diarrhea occurred between 0.1 and 5.4 mg, and only one report of diarrhea (at 24.3 mg) occurred above the 5.4 mg dose (Table 3). Additional gastrointestinal events besides diarrhea that were only reported in plecanatide-dosed subjects included nausea, abdominal discomfort, abdominal pain, and vomiting. All but one of these events occurred at the two highest doses of 24.3 and 48.6 mg. One reported event of vomiting occurred at the 8.1 mg plecanatide dose. No other trends were observed in AEs with increasing dose of plecanatide. The dose-limiting toxicity for plecanatide was therefore not diarrhea, but more closely aligned with an increased incidence of abdominal discomfort and pain at the two highest dose levels.
Overall, there were no clinically significant changes in vital signs, clinical chemistry, hematology, urinalysis, ECG, or physical exam parameters following administration of plecanatide over the entire dose range or in comparison to placebo. Overall, plecanatide was safe and well tolerated up to the highest dose administered in the study.
Discussion
This first-in-human study with plecanatide was used to assess safety, tolerability, PK, and PD of escalating single plecanatide doses compared to placebo. Overall, plecanatide was safe and well tolerated up to the highest single dose (48.6 mg) used in the study. Dosing was stopped after completion of the 48.6 mg cohort without declaring an MTD due to increasing evidence of clinically significant abdominal symptoms at the two highest dose levels.
Administration of plecanatide to volunteers resulted in no measurable plasma plecanatide concentrations in any dose cohort from 0.1 to 48.6 mg. This study utilized a validated drug assay sensitive down to a LLOQ of 10 ng/mL. Upon completion of the study, an improved assay was developed with an LLOQ of 1 ng/mL and used to re-test plasma samples from the highest dose cohort (48.6 mg dose level). Again, no plecanatide was detected in any of these samples, indicating that no measurable systemic exposure of plecanatide occurred in this single dose study.
Adverse events (AEs) in subjects receiving plecanatide (24.5 % of 53) were comparable to those receiving placebo (22.2 % of 18). Diarrhea of mild to moderate severity was the most commonly reported AE in both subjects receiving plecanatide as well as in those receiving placebo. Systemic absorption of orally administered plecanatide even at the top administered dose of 48.6 mg was below the LLOQ (1 ng/mL), suggesting that the drug acts locally in the GI tract. Notably, the majority of the TEAEs observed in this study were GI-related, which is consistent with the fact that plecanatide is a locally acting GI drug with minimal systemic exposure (Table 2).
In a similar Phase I study in healthy volunteers conducted with linaclotide, the first-in-class GC-C agonist for patients with CC and IBS-C, linaclotide was well tolerated at single oral doses up to a top dose of 3 mg, determined to be the MTD based on the incidence and severity of diarrhea [15]. Drugs acting locally in the GI tract without systemic absorption may not ever reach the typical stopping rules for dose-limiting toxicities as for other systemically absorbed drugs. This makes it difficult to truly determine MTD in the classical sense. The tolerability of GI side effects such as diarrhea rather than true safety concerns affects the limitations of drug administration in these cases. The observation that plecanatide treatment caused diarrhea in healthy volunteers could possibly be attributable to PD activity of the drug. The other treatment-related side effect observed at the two highest doses (24.3 and 48.6 mg) was nausea (5.7 %), which is likely to be due to small intestine distension [16]. Although the precise mechanism of action of plecanatide still remains to be elucidated, oral treatment with high doses of plecanatide is expected to cause excessive fluid secretion resulting in small intestine distension, which might be responsible for the nausea. However, we did not investigate the cause of nausea in this phase I study.
The apparent differences in MTD between linaclotide and plecanatide are a combination of differences in relative potencies and differences in their pharmacological actions in activating GC-C receptor. In addition, plecanatide, which is a structurally close homolog of uroguanylin, is designed to follow the physiological mechanism of uroguanylin in activating GC-C receptors. Uroguanylin shows maximum binding efficiency at a pH of approximately 5.5, which is typically observed in the upper GI, and then shows weaker binding efficiency at higher pH. By contrast, linaclotide, a homolog of E. coli heat-stable enterotoxin ST peptide, activates GC-C receptors in a pH-independent and uncontrolled manner [17]. Thus, the higher incidence of diarrhea observed in clinical evaluations with linaclotide [18] may possibly be due to excessive fluid secretion resulting from uncontrolled activation of GC-C receptors in the GI tract.
This phase I study with limited numbers of subjects was not powered to statistically assess potential PD activities of plecanatide, but the general trend in increase in BSFS scores at higher doses of plecanatide is suggestive of its pharmacological action, which is expected from an agent that activates GC-C to stimulate water secretion in the intestine. Although there was no correlation between the BSFS score and plecanatide doses, the mean BSFS score seemed to plateau at doses between 5.4 and 8.1 mg. Thus, we selected a dose range between 0.3 and 9 mg for the phase IIa clinical study in CC patients.
Conclusions
In summary, plecanatide is a member of the rapidly emerging class of GC-C agonists for treatment of functional constipating GI disorders. Overall, oral treatment with plecanatide was safe and well tolerated, with a trend in improvement of stool consistency and frequency, which is indicative of its pharmacological action. Thus, plecanatide has potential to provide relief for the multiple symptoms typically observed in patients suffering with CC and IBS-C. Current clinical trials with plecanatide in CC and IBS-C are designed to further explore its therapeutic efficacy.
References
Higgins PD, Johanson JF. Epidemiology of constipation in North America: a systematic review. Am J Gastroenterol. 2004;99:750–759.
Bharucha AE, Phillips SF. Slow transit constipation. Gastroenterol Clin North Am. 2001;30:77–95.
Pare P, Ferrazzi S, Thompson WG, Irvine EJ, Rance L. An epidemiological survey of constipation in Canada: definitions, rates, demographics, and predictors of health care seeking. Am J Gastroenterol. 2001;96:3130–3137.
Ambizas EM, Ginzburg R. Lubiprostone: a chloride channel activator for treatment of chronic constipation. Ann Pharmacother. 2007;41:957–964.
Linzess (linaclotide) Full Prescribing Information. Available at: http://www.frx.com/pi/linzess_pi.pdf. Accessed January 11, 2013.
Manabe N, Rao AS, Wong BS, Camilleri M. Emerging pharmacologic therapies for irritable bowel syndrome. Curr Gastroenterol Rep. 2010;12:408–416.
Camilleri M. Pharmacology of the new treatments for lower gastrointestinal motility disorders and irritable bowel syndrome. Clin Pharmacol Ther. 2012;91:44–59.
Harris MS, Daniels OT. Core aspects of clinical development and trials in chronic idiopathic constipation. In: Catto-Smith AG, ed. Constipation-causes, diagnosis and treatment. Rijeka: InTech; 2012:147–172.
Forte LR Jr. Uroguanylin: physiological role as a natriuretic hormone. J Am Soc Nephrol. 2005;16:291–292.
Forte LR Jr. Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol Ther. 2004;104:137–162.
Bharucha AE, Waldman SA. Taking a lesson from microbial diarrheagenesis in the management of chronic constipation. Gastroenterology. 2010;138:813–817.
Shailubhai K. Therapeutic applications of guanylate cyclase-C receptor agonists. Curr Opin Drug Discov Devel. 2002;5:261–268.
Hamra FK, Eber SL, Chin DT, Currie MG, Forte LR. Regulation of intestinal uroguanylin/guanylin receptor-mediated responses by mucosal acidity. Proc Natl Acad Sci USA. 1997;94:2705–2710.
Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–924.
Currie MG, Kurtz C, Mahajan-Miklos S, Busby RW, Fretzen A, Geis S. Effects of single dose administration of MD-1100 on safety, tolerability, exposure, and stool consistency in healthy subjects. Am J Gastroenterol. 2005;100:S328. (Abstract #894).
Barbera R, Feinle C, Read NW. Abnormal sensitivity to duedonal lipid infusion in patients with functional dyspepsia. Eur J Gastroenterol Hepatol. 1995;7:1051–1057.
Busby RW, Bryant AP, Bartolini WP, et al. Linaclotide, through activation of guanylate cyclase C, acts locally in the gastrointestinal tract to elicit enhanced intestinal secretion and transit. Eur J Pharmacol. 2010;649:328–335.
Lembo AJ, Schneier HA, Shiff SJ, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med. 2011;365:527–536.
Acknowledgments
Authors are grateful to ClinPharm Consulting, LLC (Research Triangle Park, NC, USA) for their assistance in preparation of this manuscript. This study was funded by Synergy Pharmaceuticals.
Conflict of interest
All authors are employees of Synergy Pharmaceuticals, Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shailubhai, K., Comiskey, S., Foss, J.A. et al. Plecanatide, an Oral Guanylate Cyclase C Agonist Acting Locally in the Gastrointestinal Tract, Is Safe and Well-Tolerated in Single Doses. Dig Dis Sci 58, 2580–2586 (2013). https://doi.org/10.1007/s10620-013-2684-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-013-2684-z