
Abstract
Background
Chemokine receptors are now known to play an important role in cancer growth and metastasis. However, there is little information regarding chemokine expression in gastric cancer. In this study, we examined CXCL12 expression in gastric cancer and also evaluated whether the down-regulation of CXCL12 is due to aberrant methylation of the gene.
Methods
CXCL12 expression was examined using real-time reverse-transcription polymerase chain reaction (RT-PCR), immunofluorescence, flow cytometry, and immunohistochemistry, and the methylation status of the gene was evaluated by methylation-specific PCR (MSP) in normal gastric and gastric cancer cell lines and 35 primary gastric carcinomas and corresponding nonmalignant gastric tissues.
Results
The down-regulation of CXCL12 was observed in gastric cancer cell lines and primary gastric carcinomas, while decreased expression of CXCL12 protein was significantly associated with lymph node metastasis and histological grade. And this down-regulation was found to be in accordance with aberrant methylation of the gene. Hypermethylation of the gene was observed in 65.7% (23 of 35) of the primary gastric carcinomas, while it was found in only 11.4% (4/35) of the corresponding nonmalignant tissues. Furthermore, CXCL12 expression was restored in gastric cancer cell lines after treatment with the demethylating agent, 5-aza-2′-deoxycytidine (5-Aza-dC), and demethylation of the highly metastatic cells SGC-7901 induced invasion suppression of the cells. For two CXCL12 receptors, CXCR4 and CXCR7, the mRNA levels remained almost unchanged with the 5-Aza-dC treatment.
Conclusions
Collectively, our results suggest that the aberrant methylation of CXCL12 frequently occurs in the down-regulation of CXCL12 in gastric cancers and that it may play a role in the metastasis of gastric cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chemokines have multiple roles in many kinds of physiologic processes, such as hematopoiesis, lymphocyte development, and wound healing [1]. Recent findings demonstrated that there is a close relationship between tumor cells and chemokines [2]. Cancer cell invasion and metastasis shares many similarities with the process by which leukocytes enter inflamed tissues [3]. Numerous studies indicate that chemokine receptors are expressed by tumor cells, while chemokines are expressed at organs that turn into metastatic targets. To date, the CXCL12–CXCR4 pair was found to be involved in almost all malignancies that were studied, including many solid cancers and tumors of a hematopoietic origin [4–7]. In most cases, the CXCL12–CXCR4 pair was found to be associated and/or involved with increased malignancy and metastasis, acting at many different levels [4–6, 8–15], and another CXCL12 receptor, CXCR7, was shown to promote the survival of tumor cells by preventing apoptosis, increased adhesion properties and dissemination, but did not mediate chemotaxis towards CXCL12 [16].
Recent studies have also reported that the epigenetic down-regulation of CXCL12 modulates the metastatic potential of breast, colon carcinoma, and non-small cell lung cancer [17–21]. Also, a previous study showed that many gastric cancer cell lines do not express CXCL12 [22]; however, CXCL12 protein levels were present in gastric cancer tissues by immunohistochemistry [23]. It is meaningful to understand the role of CXCL12 in gastric carcinogenesis. We investigated the expression of CXCL12 in gastric cancer and we also explored whether an epigenetic mechanism was involved in CXCL12 inactivation.
Materials and Methods
Cell Lines and Tissue Samples
Three human gastric cancer cell lines, MGC-803, BGC-823, and SGC-7901, and an immortalized normal gastric cell line, GES1, were cultured in RPMI 1640 (Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco), penicillin (100 IU/ml), and streptomycin (100 μg/ml), and incubated in a humidified incubator containing 5% CO2 at 37°C. MGC-803, BGC-823, and SGC-7901 cells were obtained from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). The GES1 cells were obtained from the Oncology Institute of China Medical University.
Tumor and corresponding nonmalignant gastric tissue specimens were obtained from 73 patients who underwent curative resection at the First Affiliated Hospital of China Medical University (Shenyang, China) between July 2009 and June 2010. None of these patients had undergone chemotherapy or radiotherapy before surgery. The local institutional review board approved our protocol for the use of patient samples; all patients provided written informed consent prior to participation in the study. Hematoxylin and eosin (H&E)-stained sections were examined histologically for the presence or absence of tumor cells. The nonmalignant gastric tissue specimens were obtained from either the opposite end of resected surgical samples or as far as possible away from the tumor. All of the macroscopically normal samples were confirmed to be normal by H&E staining.
RNA Extraction and Real-Time RT-PCR
Total RNA was isolated from cultured cells using Trizol (Invitrogen, Carlsbad, CA, USA) according to the protocol supplied by the manufacturer. Two micrograms of DNase I-treated (Fermentas, Vilnius, Lithuania) total RNA were converted to cDNA using a Reverse Transcription System kit (Fermentas). RNA was excluded in cDNA synthesis reactions as a negative control. The polymerase chain reaction (PCR) was performed in a volume of 25 μl using Maxima SYBR Green/ROX qPCR Master Mix (Fermentas) with an initial denaturing at 95°C for 10 min, followed by 40 cycles of denaturing at 95°C for 30 s, annealing for 30 s at 56°C, extension for 30 s at 72°C, and then a final extension for 10 min at 72°C. The following primers were used (5′–3′): GTC AAG CAT CTC AAA ATT CTC AAC AC (sense) and CAC TTT AGC TTC GGG TCA ATG C (antisense) for CXCL12 [24], CAG TTT CAG CAC ATC ATG GTT GG (sense) and GTG ACA GCT TGG AGA TGA TAA TGC (antisense) for CXCR4 [25], CTG CGT CCA ACA ATG AGA CCT (sense) and CCG ATC AGC CAC TCC TTG A (antisense) for CXCR7 [26], and CAT GAG AAG TAT GAC AAC AGC CT (sense) and AGT CCT TCC ACG ATA CCA AAG T (antisense) for GAPDH [27]. Relative gene expression to an internal GAPDH control and, hence, fold changes were calculated using the equation 2−ΔΔCt method [28].
Methylation-Specific PCR
The genomic DNA was prepared from cell lines and tissues by the phenol/chloroform protocol and was modified by bisulfite treatment as described previously [29]. Then, DNA (2 μg) was purified using a Wizard DNA Clean-Up System (Promega Corporation, Madison, WI, USA), precipitated with ethanol, and resuspended in 30 μl of Tris-EDTA buffer. PCR amplification was performed using 2.0 μl bisulfite-modified DNA in a volume of 50 μl containing 10× DreamTaq buffer (Fermentas), 2 mM dNTP Mix, 0.4 μM of each primer, and 1.25 U of DreamTaq (Fermentas). The primers of CXC12 used for methylation-specific PCR (MSP) are located in the promoter region and the CpG map was based on a previous study [18]. The primers for the methylated CXCL12 CpG island were 5′-GGA GTT TGA GAA GGT TAA AGG TC-3′ (sense) and 5′-TTA ACG AAA AAT AAA AAT AGA CGA T-3′ (antisense). The primers for the unmethylated CXCL12 CpG islands were 5′-GAG TTT GAG AAG GTT AAA GGT TGG-3′ (sense) and 5′-TAA CAA AAA ATA AAA ATA CAA CAA T-3′ (antisense). The PCR conditions were as follows: 95°C for 10 min, followed by 39 cycles of denaturing at 95°C for 30 s, annealing for 30 s at 60°C, extension for 30 s at 72°C, and then a final extension for 10 min at 72°C. CpGenome Universal Methylated and Unmethylated DNA (Chemicon, Temecula, CA, USA) was used as a positive control for the methylated and unmethylated genes, respectively. The amplification products were separated on 2.5% agarose gels.
Immunofluorescence
Indirect immunofluorescences were done on CXCL12 cells grown in 24-well plates (Costar Corp, Corning, NY, USA). Cells were fixed in 4% paraformaldehyde at 25°C for 15 min and permeabilized with 0.2% Triton X-100, and then washed three times with PBS. Cells were incubated with 25 μg/ml murine anti-human CXCL12 monoclonal antibody (MAB350 R&D Systems, Minneapolis, MN, USA) or an isotype control (MAB002 R&D Systems) overnight, followed by incubation with a 1:100 dilution of goat anti-murine TRICT-conjugated IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 h in the dark. Nuclei were visualized with Hochest33258 counterstain and examined using a fluorescence microscope (Olympus BX-40).
Flow Cytometry
The cells were fixed in 4% paraformaldehyde for 15 min on ice and permeabilized with 0.1% saponin. Then, they were washed and incubated with 25 μg/ml murine anti-human CXCL12 monoclonal antibody for 1 h on ice. After being washed three times, bound primary antibody were detected by incubation with FITC-labeled anti-murine IgG (Santa Cruz Biotechnology) for 1 h on ice in the dark. Cells were washed extensively, resuspended in PBS, and analyzed by flow cytometry (FACScan, Becton Dickinson, Mountain View, CA, USA). Murine IgG served as an isotype-negative control. The data were expressed as a ratio between the mean fluorescence intensity (MFI) of specific antibody and the MFI of the relative isotype control. The results were obtained from three independent experiments for each cell line.
Immunohistochemistry
For the immunohistochemical examination, we followed the normal protocol in preparing the tissues and the epitope retrieval was performed in citrate buffer (pH 6) using a microwave for 2 min at 100°C. After blocking endogenous peroxidase with 3% H2O2, we followed all of the protocols according to the Ultra Sensitive S-P secondary antibody kit (KIT-9701, Maixin Bio, Fuzhou, China). The primary antibody or an isotype control was incubated overnight at 4°C at 1.25 μg/ml. Finally, detection was carried out with the DAB Kit (DAB-003, Maixin Bio, Fuzhou, China) as described by the manufacturer.
Following immunostaining, the slides were first analyzed manually. Any cytoplasmic staining for CXCL12 was considered to be positive. The immunostaining results were evaluated by defining a threshold of positive staining for all sections before automated processing. The immunohistochemical parameter assessed in the area detected was the mean density and the intensity was averaged from five fields of view with a magnification of 400×. This was performed using software Image-Pro Plus 6.0 (Media Cybernetics, Bethesda, MD, USA).
Treatment of Cells with 5-Aza-dC
Three tumor cell lines were incubated in culture media with 5 μmol/l of the demethylating agent 5-aza-2′-deoxycytidine (5-Aza-dC) (Sigma Chemical Co., St. Louis, MO, USA) for 3 days using daily media changes. Cells were harvested and RNA and DNA were extracted on day 3.
Transwell Chamber Invasion Assay
The transwell chamber (Corning Life Sciences, Corning, NY, USA) containing an 8-μm pore size polycarbonate membrane filter was coated with a Matrigel (BD Biosciences, San Jose, CA, USA) and inserted in a 24-well culture plate. SGC-7901 cells and SGC-7901 cells incubated with 5-Aza-dC for 3 days were separately detached from the tissue culture plates, washed, and plated at the density of 5 × 104 per upper well in 200 μl of culture medium (RPMI 1640, 1% FBS), and the lower chamber was filled with 500 μl of medium (RPMI 1640, 20% FBS). After being recultured with 5% CO2 at 37°C for 24 h, the noninvading cells with Matrigel matrix were removed from the upper surface of the membrane by scrubbing with a cotton-tipped swab. Cells on the lower surface of the filter were fixed for 30 min in methanol and glacial acetic acid mixture (3:1), air-dried briefly, and stained with Giemsa. The number of invaded cells was counted from three preselected microscopic fields at 200× magnification; all experiments were performed in triplicate.
Statistical Analysis
Statistical analysis was performed using the statistical software package SPSS version 12.0 (LEAD Technologies Inc., Chicago, IL, USA). Each data point shows a representative result from a series of experiments done on at least three independent occasions. A P-value < 0.05 was considered to be statistically significant. Significance between controls and treated cell lines were calculated by Student’s t-test. Significance between controls in different cell lines was calculated by one-way analysis of variance (ANOVA). The correlations of the CXCL12 protein level immunostained in gastric cancers and clinicopathological characteristics were assessed by the Kruskal–Wallis test and the Mann–Whitney test. The correlations of the methylation alterations of the CXCL12 gene with clinicopathological parameters were analyzed by the Chi-square test and Fisher’s exact test.
Results
Down-Regulation of CXCL12 in Gastric Cancer Cell Lines
We first sought to define the CXCL12 expression pattern in three gastric cancer cell lines and GES1. As an initial step, we performed real-time RT-PCR to assess the mRNA expression of CXCL12. Quantitative RT-PCR revealed a considerable down-regulation of CXCL12 in the tumor cell lines MGC-803, BGC-823, and SGC-7901 compared with the normal cell line GES1 (P = 0.00) (Fig. 1a).

Decreased expression of CXCL12 in gastric cancer cell lines. a Quantitative reverse-transcription polymerase chain reaction (RT-PCR) analyses on the expression of CXCL12 in GES1 and three gastric cancer cell lines (MGC-803, BGC-823, and SGC-7901). The columns indicate the mean CXCL12 expression levels normalized to GAPDH expression. *P < 0.05 compared with GES1. b The GES1 cells permeabilized and immunostained with antibody to CXCL12 (red) were characterized by perinuclear cytoplasmic immunofluorescence. Cell nuclei were visualized with Hochest33258 (blue). CXCL12 signals were not detected on MGC-803, BGC-823, and SGC-7901. Original magnification 400×. c Intracellular protein expression of CXCL12 was detected in permeabilized GES1 cells using flow cytometry (solid histograms). The clear histograms correspond with immunostaining with the appropriate isotype control IgG, whereas lower CXCL12 expression levels were detected in MGC-803, BGC-823, and SGC-7901. The columns indicate the mean fluorescence intensity (MFI) of CXCL12 protein expression levels. *P < 0.05 compared with GES1
We next analyzed CXCL12 protein expression using two parallel approaches, flow cytometry and immunofluorescence, to determine if dysregulated protein expression was paralleled in those cell lines. Perinuclear cytoplasmic CXCL12 immunofluorescence was observed in GES1, but not in other cancer cell lines. No specific immunofluorescences were observed using murine isotype control antibodies for CXCL12 (Fig. 1b). To detect intracellular CXCL12 protein using flow cytometry, permeabilized GES1 cells were immunostained with monoclonal antibodies and were shown to express CXCL12. However, lower CXCL12 expression levels were detected in MGC-803, BGC-823, and SGC-7901 (P = 0.00) (Fig. 1c).
Correlation Between the Down-Regulation of CXCL12 and Clinical Factors in Primary Gastric Carcinomas
CXCL12 protein was detected in the cytoplasm of normal gastric epithelial cells and well-differentiated gastric cancer cells, especially in the most differentiated surface gastric epithelial cells, whereas lower CXCL12 expressions were detected in the less differentiated epithelial cells distant from the mucosal side and the less differentiated gastric cancer cells (Fig. 2).

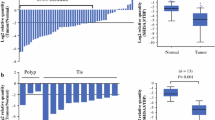
Immunohistochemical analysis of CXCL12 protein in primary gastric carcinomas and corresponding nonmalignant tissues. Representative tissue samples stained with an anti-CXCL12 antibody demonstrate weak staining in cancerous tissues (d, e, f), but strong staining in normal tissues (a, c). CXCL12 was strongly expressed in the cytoplasm of the most differentiated surface gastric epithelial cells, but moderate in the less differentiated epithelial cells far from the mucosal side (c). CXCL12 expression by primary gastric carcinoma cells was variable and reduced, CXCL12-specific staining was strong in well-differentiated gastric cancer cells (d), while there was weak or no staining in less-differentiated gastric cancer cells (e, f). Immunoglobulin G (IgG) control images for CXCL12 (b) were shown negatively. The images are shown at 100× magnification, with the boxed areas indicating the 400× images
Quantitative analysis of the CXCL12 protein by the mean density of staining in 73 pairs of primary gastric carcinomas and corresponding nonmalignant gastric tissue revealed significantly (P = 0.00) lower levels in gastric carcinoma tissues (median = 0.006150, range = 0.000090–0.147000) than in normal gastric tissues (median = 0.0258000, range = 0.000050–0.227000). Furthermore, lower protein expression levels of CXCL12 in tumors were related to lymph node metastasis (P = 0.04) and histological grade (P = 0.00) (Table 1).
CXCL12 Silencing by DNA Methylation
The MGC-803 and SGC-7901 gastric cancer cell lines only showed methylated bands (hypermethylated, both alleles methylated), which were in agreement with the observed low levels of CXCL12 expression. In contrast, the GES1 cell line expressing high CXCL12 levels showed unmethylated bands (unmethylated, neither allele is methylated) and the BGC-823 cell line showed both methylated and unmethylated bands (partially methylated, only one allele methylated) (Fig. 3a).

CXCL12 methylation analyses in cell lines and tumors and their corresponding nonmalignant gastric tissues by methylation-specific PCR (MSP). a Methylated bands were detected in MGC-803 and SGC-7901. Unmethylated bands were detected in GES1, and both methylated and unmethylated bands were detected in BGC-823. b Representative examples of the methylation analysis of CXCL12 in tumors and their corresponding nonmalignant gastric tissues. M size marker, N nonmalignant gastric tissue, T tumor, u amplified product with primers recognizing an unmethylated sequence, m amplified product with primers recognizing a methylated sequence, Pos positive control
We then examined the methylation status of the CXCL12 promoter in 35 primary gastric carcinomas as well as nonmalignant gastric tissues from the same individuals by MSP (Fig. 3b). Hypermethylation of the CXCL12 gene was detected in 23 (65.7%) of the 35 primary gastric carcinomas, while hypermethylation in nonmalignant gastric tissue was only found in 4 cases (11.4%). And CXCL12 was partially methylated in 8 cases (22.9%) and non-methylated in 4 cases (11.4%) in primary gastric tumors and 4 cases (11.4%) of partially methylation and 27 cases (77.1%) of non-methylation in noncancerous tissues, respectively.
We also examined the relationship between gastric tumor CXCL12 methylation status and the clinicopathologic features of the patients, including age, gender, depth of invasion, lymph node status, histological grade, and pathologic staging of the tumor, but no correlation with any of the clinicopathologic features of the patients was found (Table 1).
Reactivation of CXCL12 Expression After Treatment with 5-Aza-dC
To confirm that aberrant methylation was responsible for silencing CXCL12 expression, we treated the MGC-803, BGC-823, and SGC-7901 gastric cancer cell lines with the demethylating agent 5-Aza-dC. The methylation status of CXCL12 of gastric cancer cells was modified from methylated to unmethylated by the 5-Aza-dC treatment (Fig. 4d). The expression of CXCL12 mRNA (Fig. 4a) and protein (Fig. 4b, c) were restored in MGC-803 (P = 0.00; P = 0.00) and SGC-7901 (P = 0.00; P = 0.00) cells, but not in BGC-823 (P = 0.89; P = 0.12) cells. The mRNA levels of the two receptors CXCR4 and CXCR7 in MGC-803 (P = 0.12; P = 0.30) and SGC-7901 (P = 0.45; P = 0.44) cells and the CXCR7 mRNA levels in BGC-823 (P = 0.40) cells remained almost unchanged with the 5-Aza-dC treatment, except for CXCR4 mRNA in BGC-823 cells (P = 0.01) (Fig. 4a).

Demethylation analysis using gastric cancer cell lines. a CXCL12 and its two receptors CXCR4 and CXCR7 mRNA expression evaluation after the 5-aza-2′-deoxycytidine (5-Aza-dC) treatment in MGC-803, BGC-823, and SGC-7901 cell lines using real-time RT-PCR. *P < 0.05 compared with cells before the 5-Aza-dC treatment. b CXCL12 fluorescent immunocytochemical signals were re-visualized in MGC-803, BGC-823, and SGC-7901 cells after 5-Aza-dC exposure. 400× original magnification. c Effect of the 5-Aza-dC treatment on CXCL12 protein expression analyzed by flow cytometry in MGC-803, BGC-823, and SGC-7901 cell lines. The clear histograms represent cell lines before the 5-Aza-dC treatment, whereas the solid histograms indicate corresponding cells after the treatment. The columns indicate the MFI of CXCL12 protein expression levels. *P < 0.05 compared with cells before the 5-Aza-dC treatment. d CXCL12 MSP indicating the presence of unmethylated bands of CXCL12 in MGC-803, BGC-823, and SGC-7901 cell lines treated with 5 μmol/l 5-aza for 3 days. M size marker, u amplified product with primers recognizing an unmethylated sequence, m amplified product with primers recognizing a methylated sequence
Reactivation of CXCL12 Expression with 5-Aza-dC Inhibits the Invasion Ability of Highly Metastatic SGC-7901 Cells
To further examine whether the reactivation of CXCL12 expression can regulate gastric cancer invasion, we analyzed the invasion capability of the highly metastatic SGC-7901 cells using the methods described above. The number of SGC-7901 cells in the untreated group that migrated through the membrane was 140.00 ± 12.12. The number of invading cells was significantly decreased when SGC-7901 cells were treated with 5-Aza-dC (63.67 ± 6.03). A significant reduction in the number of invasive cells was observed for 24 h when the cells were treated with 72 h of 5-Aza-dC exposure compared to the control (P = 0.001) (Fig. 5).

Effect of 5-Aza-dC on SGC-7901 cells invasion. The transwell assay was performed to assess the effect on cell invasion. Cells were treated or untreated with 5 M 5-Aza-dC. Representative photographs of treated and untreated cells are presented (200× magnification). The columns indicate the number of cells invaded at the 24-h time point. *P < 0.05 compared with untreated SGC-7901 cells. The values represent the mean values ± standard deviation (SD)
Discussion
Various types of cancer cells express chemokine receptors and the chemokines may play a role in cancer progression and/or organ-selective metastasis. It is assumed that disseminated tumor cells expressing chemokine receptors invade the circulation and are then attracted and arrested by the corresponding ligand. The specific metastatic sites to which tumor cells preferred to metastasize expressed more chemokines, and these chemokines are then able to induce the migration of tumor cells [10–12, 30–32]. The ability of a specific chemokine to act on chemokine receptor-expressing tumor cells and to support their directionality requires that chemokine-induced cellular changes occurring in the tumor cells would culminate into motility in response to chemokine gradients. Solid evidence to such a mechanism was provided by the study of Müller et al., demonstrating that a highly potent functional axis exists between CXCL12 and its CXCR4 receptor in breast cancer metastasis [30]. In addition to this role, CXCR4 signaling is also a key regulator of organogenesis as well as lymphopoiesis and myelopoiesis [33, 34]. Previous studies have defined the co-expression of both CXCR4 and CXCL12 by the cells of the human intestinal epithelium [35], and, also, many studies reported the different expression levels of CXCL12 in the cancer cells and tissues [36, 37].
In this study, we demonstrated that lower expression levels of CXCL12 mRNA and protein occur in gastric cancer cell lines, which is similar to the results found by Yasumoto et al. [22], who showed the role of CXCR4-expressing gastric cancer cells in peritoneal carcinomatosis. Also, these investigators found no mRNA and protein expression of CXCL12 in any gastric cancer cell lines, which definitely showed no autocrine of CXCL12 in the gastric cancer cells. Moreover, we also found that the expression of CXCL12 was frequently reduced in gastric primary carcinomas when compared to corresponding normal gastric tissues. Other researchers found the same phenomenon in colon cancer, mammary carcinoma, and non-small lung cancer, and the CpG islands of the CXCL12 gene was hypermethylated [17–20]. One of our objectives was to verify whether the aberrant methylation findings were in gastric carcinomas. We evaluated the CpG islands of the CXCL12 gene that had already been analyzed by another group that found DNA methylation in colon cancer and mammary carcinoma [17, 18, 20]. In our research, the CpG islands of the CXCL12 gene was hypermethylated in 2 of 3 gastric cancer cells, but not in GES1 cells, and as high as 65.7% in the 35 primary gastric tumors, whereas only 11.4% hypermethylation of CXCL12 was observed in corresponding normal gastric tissues. To further explore the DNA methylation, we treated cancer cells with 5-Aza-dC, a DNA methyltransferase inhibitor, which was sufficient to cause demethylation of the promoter region and reactivate the expression of the hypermethylated silenced gene [38]. After 5-Aza-dC treatment, we observed a complete reversal of CXCL12 mRNA and protein expression in MGC-803 and SGC-7901 cells. As shown in this study, the action of 5-Aza-dC on the gastric cancer cell lines MGC-803 and SGC-7901 resulted in the demethylation of the CXCL12 gene, accompanied by the up-regulation of mRNA and protein. This confirmed that 5-Aza-dC regulated the transcription of the CXCL12 gene. In addition, the effect of 5-Aza-dC on the invasion ability of the highly metastatic gastric cancer cells SGC-7901 was demonstrated in vitro using the transwell assay. Our data suggest that the invasion ability suppression effect of 5-Aza-dC may result from the demethylation and reactivation of CXCL12. The down-regulation of CXCL12 was correlated with its promoter methylation in both gastric cancer cell lines and gastric tumor tissues. These results suggest that the aberrant methylation of CXCL12 might be involved in the metastasis of gastric cancer.
In this study, we found that the CXCL12 was frequently down-regulated by the methylation of its promoter in gastric cancer. Loss of CXCL12 with maintained expression of CXCR4 imparts to metastatic cancer cells a phenotype similar to that of circulating highly migratory leukocytes and lymphocytes [39]. Our data have also shown that lower CXCL12 protein levels in gastric tumors were related to lymph nodes metastasis, suggesting that carcinoma cells with no autocrine of CXCL12 may have a selective advantage to receive endocrine CXCL12 signals, promoting their exit and driving more active metastasis to ectopic sources of the CXCR4 ligand [17, 18]. In our study, we also found that the CXCL12 expression was restored by 5-Aza-dC treatment with no changes of the two cognate receptors CXCR4 and CXCR7, which is a new CXCL12 receptor that has been recently identified [16, 40]. This finding indicates that the CXCL12 changes in the gastric cancer cells could not affect its receptors, which reinforces the notion that the endogenous and exogenous CXCL12 play different roles. Endogenous CXCL12, in marked contrast to exogenous ligand, inhibits tumor metastasis through increased anoikis [39]. Drury et al. found that CXCL12 expression induced apoptosis specifically in nonadherent colorectal carcinoma cells by increasing anoikis via the activation of a Bim-mediated intrinsic apoptotic pathway [41].
In our research, the CpG islands of the CXCL12 gene were partially methylated in BGC-823 and were not hypermethylated in 13 of the 35 primary gastric carcinomas, which suggested to us that the aberrant methylation of the gene might not be the only reason to down-regulate the expression of CXCL12. An understanding of the precise mechanism of the down-regulation of endogenous CXCL12 in these gastric cancer cells needs to be explored.
Conclusions
In conclusion, we examined endogenous CXCL12 expression in gastric cancer cells and tissues. We also explored whether the decreased CXCL12 expression occurred as a result of aberrant methylation of the gene. We found that the CXCL12 was frequently down-regulated by the methylation of its promoter in gastric cancers, which suggests that endogenous CXCL12, in marked contrast to exogenous ligand, inhibits tumor metastasis in gastric cancer, which was the first research to explore the mechanism of endogenous CXCL12 down-regulation in gastric cancer. Our results, together with recent findings, emphasize the importance of the CXCL12-CXCR4 signaling axis in an organ-specific pattern of metastasis. Targeting CXCL12 signal may also be a novel and efficient strategy for treating human gastric cancers.
Abbreviations
- 5-Aza-dC:
-
5-Aza-2′-deoxycytidine
- RT-PCR:
-
Reverse-transcription polymerase chain reaction
- MSP:
-
Methylation-specific PCR
References
Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi:10.1016/S1074-7613(00)80165-X.
Scotton CJ, Wilson JL, Milliken D, Stamp G, Balkwill FR. Epithelial cancer cell migration: a role for chemokine receptors? Cancer Res. 2001;61:4961–4965.
Koizumi K, Hojo S, Akashi T, Yasumoto K, Saiki I. Chemokine receptors in cancer metastasis and cancer cell-derived chemokines in host immune response. Cancer Sci. 2007;98:1652–1658. doi:10.1111/j.1349-7006.2007.00606.x.
Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006;25:357–371. doi:10.1007/s10555-006-9003-5.
Balkwill F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol. 2004;14:171–179. doi:10.1016/j.semcancer.2003.10.003.
Ratajczak MZ, Zuba-Surma E, Kucia M, Reca R, Wojakowski W, Ratajczak J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia. 2006;20:1915–1924. doi:10.1038/sj.leu.2404357.
Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi:10.1038/nrc1388.
Zlotnik A. Chemokines in neoplastic progression. Semin Cancer Biol. 2004;14:181–185. doi:10.1016/j.semcancer.2003.10.004.
Salcedo R, Oppenheim JJ. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation. 2003;10:359–370. doi:10.1038/sj.mn.7800200.
Luker KE, Luker GD. Functions of CXCL12 and CXCR4 in breast cancer. Cancer Lett. 2006;238:30–41. doi:10.1016/j.canlet.2005.06.021.
Wang J, Loberg R, Taichman RS. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006;25:573–587. doi:10.1007/s10555-006-9019-x.
Dewan MZ, Ahmed S, Iwasaki Y, Ohba K, Toi M, Yamamoto N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother. 2006;60:273–276. doi:10.1016/j.biopha.2006.06.004.
Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi:10.1182/blood-2005-08-3182.
Epstein RJ. The CXCL12-CXCR4 chemotactic pathway as a target of adjuvant breast cancer therapies. Nat Rev Cancer. 2004;4:901–909. doi:10.1038/nrc1473.
Zlotnik A. Involvement of chemokine receptors in organ-specific metastasis. Contrib Microbiol. 2006;13:191–199. doi:10.1159/000092973.
Burns JM, Summers BC, Wang Y, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–2213. doi:10.1084/jem.20052144.
Wendt MK, Cooper AN, Dwinell MB. Epigenetic silencing of CXCL12 increases the metastatic potential of mammary carcinoma cells. Oncogene. 2008;27:1461–1471. doi:10.1038/sj.onc.1210751.
Wendt MK, Johanesen PA, Kang-Decker N, Binion DG, Shah V, Dwinell MB. Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene. 2006;25:4986–4997. doi:10.1038/sj.onc.1209505.
Suzuki M, Mohamed S, Nakajima T, et al. Aberrant methylation of CXCL12 in non-small cell lung cancer is associated with an unfavorable prognosis. Int J Oncol. 2008;33:113–119.
Ramos EA, Camargo AA, Braun K, et al. Simultaneous CXCL12 and ESR1 CpG island hypermethylation correlates with poor prognosis in sporadic breast cancer. BMC Cancer. 2010;10:23. doi:10.1186/1471-2407-10-23.
Zhou W, Jiang Z, Liu N, et al. Down-regulation of CXCL12 mRNA expression by promoter hypermethylation and its association with metastatic progression in human breast carcinomas. J Cancer Res Clin Oncol. 2009;135:91–102. doi:10.1007/s00432-008-0435-x.
Yasumoto K, Koizumi K, Kawashima A, et al. Role of the CXCL12/CXCR4 axis in peritoneal carcinomatosis of gastric cancer. Cancer Res. 2006;66:2181–2187. doi:10.1158/0008-5472.CAN-05-3393.
Ishigami S, Natsugoe S, Okumura H, et al. Clinical implication of CXCL12 expression in gastric cancer. Ann Surg Oncol. 2007;14:3154–3158. doi:10.1245/s10434-007-9521-6.
Zieker D, Königsrainer I, Traub F, et al. PGK1 a potential marker for peritoneal dissemination in gastric cancer. Cell Physiol Biochem. 2008;21:429–436. doi:10.1159/000129635.
Zieker D, Königsrainer I, Tritschler I, et al. Phosphoglycerate kinase 1 a promoting enzyme for peritoneal dissemination in gastric cancer. Int J Cancer. 2010;126:1513–1520. doi:10.1002/ijc.24835.
Raggo C, Ruhl R, McAllister S, et al. Novel cellular genes essential for transformation of endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 2005;65:5084–5095. doi:10.1158/0008-5472.CAN-04-2822.
Richard CL, Tan EY, Blay J. Adenosine upregulates CXCR4 and enhances the proliferative and migratory responses of human carcinoma cells to CXCL12/SDF-1alpha. Int J Cancer. 2006;119:2044–2053. doi:10.1002/ijc.22084.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi:10.1006/meth.2001.1262.
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826.
Müller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi:10.1038/35065016.
Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–1837.
Oonakahara K, Matsuyama W, Higashimoto I, Kawabata M, Arimura K, Osame M. Stromal-derived factor-1alpha/CXCL12-CXCR 4 axis is involved in the dissemination of NSCLC cells into pleural space. Am J Respir Cell Mol Biol. 2004;30:671–677. doi:10.1165/rcmb.2003-0340OC.
Nagasawa T, Kikutani H, Kishimoto T. Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc Natl Acad Sci USA. 1994;91:2305–2309.
Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi:10.1038/31269.
Dwinell MB, Eckmann L, Leopard JD, Varki NM, Kagnoff MF. Chemokine receptor expression by human intestinal epithelial cells. Gastroenterology. 1999;117:359–367. doi:S0016508599001535.
Yoshitake N, Fukui H, Yamagishi H, et al. Expression of SDF-1 alpha and nuclear CXCR4 predicts lymph node metastasis in colorectal cancer. Br J Cancer. 2008;98:1682–1689. doi:10.1038/sj.bjc.6604363.
Brand S, Dambacher J, Beigel F, et al. CXCR4 and CXCL12 are inversely expressed in colorectal cancer cells and modulate cancer cell migration, invasion and MMP-9 activation. Exp Cell Res. 2005;310:117–130. doi:10.1016/j.yexcr.2005.07.006.
Meng CF, Zhu XJ, Peng G, Dai DQ. Role of histone modifications and DNA methylation in the regulation of O6-methylguanine-DNA methyltransferase gene expression in human stomach cancer cells. Cancer Invest. 2010;28:331–339. doi:10.3109/07357900903179633.
Wendt MK, Drury LJ, Vongsa RA, Dwinell MB. Constitutive CXCL12 expression induces anoikis in colorectal carcinoma cells. Gastroenterology. 2008;135:508–517. doi:10.1053/j.gastro.2008.05.033.
Balabanian K, Lagane B, Infantino S, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280:35760–35766. doi:10.1074/jbc.M508234200.
Drury LJ, Wendt MK, Dwinell MB. CXCL12 chemokine expression and secretion regulates colorectal carcinoma cell anoikis through bim-mediated intrinsic apoptosis. PLoS One. 2010;5:e12895. doi:10.1371/journal.pone.0012895.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant number: 30572162).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhi, Y., Chen, J., Zhang, S. et al. Down-Regulation of CXCL12 by DNA Hypermethylation and Its Involvement in Gastric Cancer Metastatic Progression. Dig Dis Sci 57, 650–659 (2012). https://doi.org/10.1007/s10620-011-1922-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-011-1922-5