
Abstract
Purpose
Not only is the expression of CXCR4 on breast cancers a key determinant of tumor metastasis, CXCL12 exhibiting peak levels of constitutive expression in organs representing the first destinations of cancer metastasis, but is proposed to be also essential for the organ-specific metastatic process.
Methods
In this study, the expressions of CXCR4 and CXCL12 were investigated using quantitative RT-PCR and immunohistochemistry in samples of 63 primary breast carcinomas and 20 normal breast tissues. Using methylation-specific PCR, we also analyzed the methylation status of CXCL12.
Results
Both up-regulation of CXCR4 and down-regulation of CXCL12 were observed in primary breast carcinomas. Over-expression of CXCR4 mRNA was significantly related to lymph node metastasis status and strong Her-2 expression, while decreased expression of CXCL12 mRNA was significantly associated with positive lymph node metastasis and estrogen receptor negativity. Methylation-specific PCR showed that 52.4% of breast tumors were hypermethylated in the CXCL12 promoter region. The expression levels of DNA methyltransferase (DNMT) 1 and DNMT3B were significantly higher in the CXCL12-methylated breast carcinomas than in the CXCL12-unmethylated ones.
Conclusions
In summary, DNA hypermethylation of CXCL12 plays an important role in the down-regulation of CXCL12 expression in breast carcinomas. Cancer cells lacking expression of CXCL12, but maintaining over-expression of CXCR4, can selectively spread to target organs in which the ligand is highly secreted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chemokines are small, secreted chemotactic proteins which are now the largest known cytokine family. All scientists and clinicians in oncology-related fields are now aware of their crucial roles at all stages of neoplastic transformation and progression (Arya et al. 2007). Among chemokines and their receptors, the CXCL12/CXCR4 system has been demonstrated to regulate several key processes in a wide variety of cancers. Particularly, studies have identified its pivotal role in the directional migration of cancer cells during the metastatic process (Arya et al. 2007; Zlotnik 2006). As we know, primary breast carcinoma is characterized by a distinctive pattern of metastases, which does not correlate with patterns of blood-flow, but suggests preferential homing, adhesion, survival, and proliferation in specific organs and tissues (Luker and Luker 2006). CXCL12, formerly known as stromal cell-derived factor-1 (SDF-1), exhibits peak levels of expression in a variety of tissues representing the first destinations of breast cancer metastasis such as lung, liver, lymph node, bone marrow, and adrenal gland (Müller et al. 2001). On the other hand, its exclusive receptor CXCR4, a seven-membrane-spanning G-protein coupled receptor (GPCR), is highly expressed in human breast cancer cells, malignant breast tumors, and metastases (Müller et al. 2001; Shim et al. 2006). CXCR4 signaling in response to CXCL12 mediates actin polymerization and pseudopodia formation, and subsequently induces chemotactic and invasive responses. In vivo, neutralizing the interactions of CXCL12/CXCR4 significantly impairs metastasis of breast cancer cells to regional lymph nodes and lung (Müller et al. 2001). These data have identified the key function of the CXCL12/CXCR4 signaling axis in the metastatic process of breast cancer.
However, it has been recently demonstrated that CXCL12 is absent in several colonic cancer cell lines and primary carcinoma tissues (Wendt et al. 2006). Furthermore, re-establishing endogenous expression of CXCL12 in colonic carcinoma cells profoundly reduced in vivo metastatic tumor formation (Wendt et al. 2006). Wendt et al. first proposed that silencing the ligand arm of this signaling axis changes the homeostatic autocrine and paracrine CXCL12/CXCR4 signaling to a strictly endocrine communication arc that facilitates metastasis of colonic carcinomas (Wendt et al. 2006). They also found similar results in breast carcinoma cell lines (Wendt et al. 2008). However, the expression patterns of the CXCL12/CXCR4 signaling axis and their relationship with clinicopathological data in primary tumors, especially in breast carcinoma, is largely unknown.
In primary human breast cancer, aberrant methylation has been revealed to be associated with the inactivation of critical tumor suppressor and growth regulatory genes, such as cell cycle regulating (p16, 14–3–3σ), steroid receptor (ERα, PR, RARβ2), tumor susceptibility (BRCA1), carcinogen detoxification (GSTP1), cell adhesion (E-cadherin) and inhibitors of matrix metalloproteinases genes (TIMP-3) (Yang et al. 2001). But few findings have been obtained on proto-oncogenes. An online search for CpG islands in the 5′ region of the CXCL12 gene revealed a typical CpG island on the start site of the transcription. Importantly, this region of CXCL12 lacks a true TATA-box. It indicated that hypermethylation of the promoter regions of CXCL12 likely plays an important role in the regulation of their mRNA levels in malignant cancers. Wendt et al. (2006, 2008) demonstrated that hypermethylation of the CXCL12 promoter region was evident in 62% of 21 colonic carcinomas, 40% of 15 breast carcinomas, but not in any of normal corresponding tissues. In the present study, we enlarged the group of detected samples, and emphasized on their relationship analysis of CXCL12 expression/methylation status with clinicopathological features in primary breast carcinomas. In addition, given that over-expression of the enzymes catalyzing DNA methylation may be a prerequisite for abnormal DNA methylation (Robertson 2001), we have also determined the correlation of DNA methyltransferases (DNMTs) expression levels, especially DNMT1, 3A, and 3B, with CXCL12 methylation status in primary breast carcinomas.
Materials and methods
Clinical tissue samples
Primary breast carcinoma samples (n = 63) and normal breast tissues (n = 20) were obtained from patients diagnosed with T1–T3, N0–N2 (stage I–IIIA) breast carcinoma, treated with curative resectional surgery between June 2004 and March 2006 in Shandong Cancer Hospital and Institute (Jinan, China). Nine patients had stage I, 25 had stage IIA, 13 had stage IIB, and 16 had stage IIIA disease. They were classified according to the WHO classification (World Health Organisation 1981). These patients had neither chemotherapy nor radiotherapy before operation. The mean age of the patients was 53.0 years (range 32–80). There were 34 node-negative and 29 node-positive patients (12 with one to three positive nodes, and 17 with more than three positive nodes). Histological subtypes included 43 invasive ductal, 6 invasive lobular, 6 mixed type (ductal/lobular), 7 mucinous carcinomas and 1 ductal in situ carcinoma. All the breast tissue samples were collected with the informed consent of all patients and approval of the ethics committee of Shandong Cancer Hospital. The specimens collected were snap-frozen in liquid nitrogen and kept at −80°C.
RNA extraction and reverse transcription
Total RNA was extracted from frozen tissues using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. RNA was treated with DNase and purified using the RNeasy mini kit (Qiagen, Hilden, Germany). cDNA synthesis was conducted using the high capacity cDNA archive kit (Applied Biosystems) according to the manufacturer’s instructions.
Semi-quantitative RT-PCR
Primer sequences for CXCL12, CXCR4 (Kim et al. 2006), DNMTs (Girault et al. 2003; Kwon et al. 2007) and the other primers used in this study, annealing temperatures, and the expected PCR product sizes are summarized in Table 1. GAPDH, as an internal control, was also amplified in the same PCR reactions. Cycling conditions where denaturation at 94°C for 5 min, then 35 cycles of PCR (94°C, 30 s; 60°C, 30 s; and 72°C, 30 s), followed by a final extension of 72°C for 10 min. The amplification products were resolved by 3% agarose gel electrophoresis and visualized by ethidium bromide staining.
Quantitative real-time PCR
Quantitative real-time RT-PCR was carried out using the specific primers as mentioned in Table 1. GAPDH mRNA was also amplified as an internal control. Briefly, 2 μl of cDNA was used to determine gene expression levels by the ABI PRISM® 7000HT Sequence Detection Systems (Applied Biosystems, USA). It was amplified in the 25 μl reaction including 2xSYBR Premix Ex TaqTM 12.5 μl (Takara, Japan), 10 μM of each primer 0.5 μl, 50xROX Reference Dye 0.5 μl and H2O 9 μl. Cycling conditions included a hot start (95°C, 15 s), followed by 40 cycles of 95°C, 10 s and 60°C, 40 s. After control for equal PCR efficiency of target genes and internal controls, relative gene expressions were presented with the comparative Ct method of quantification (Livak and Schmittgen 2001).
DNA preparation and sodium bisulfate treatment
Genomic DNA was prepared from frozen tumor tissues by standard SDS/proteinase K digestion followed by phenol/chloroform extraction and ethanol precipitation. For DNA methylation analysis, sodium bisulfite modification of genomic DNA was performed, as described previously with minor modifications (Grunau et al. 2001; Jiang et al. 2006). Briefly, 1 μg of genomic DNA was at 42°C for 30 min, in a total volume of 50 μl containing NaOH (final concentration 0.2 M). After the addition of 30 μl freshly prepared 10 mM hydroquinone (Sigma, USA), and 520 μl of 3 M sodium bisulfite (Sigma, USA) at pH 5.0, the samples were incubated for 16 h at 55°C in the dark. Afterward, modified DNA was purified using Wizard DNA clean-up resin (Promega A7280, Madison, USA), and DNA was desulfonated in 0.3 M NaOH at 37°C for 15 min. The converted DNA was precipitated at −20°C, overnight in ice-cold 100% ethanol containing ammonium acetate (final concentration, 3 M) and 10 μg of glycogen.
Methylation-specific PCR
Bisulfite-modified DNA was amplified with primers specific for methylated or unmethylated sequences. The methylated- and unmethylated-specific primers (Wendt et al. 2006) covered the transcription start site of CXCL12 (Table 1). The PCR mixture contained 2× GC buffer 5 μl (Takara, Japan), 4 × 10 mM dNTP 0.4 μl, 10 μM of each primer 0.5 μl, 5 U/μl (Takara, Japan), LA Taq DNA polymerase 0.1 μl (Takara, Japan), and 2 μl bisulfite-modified DNA. Amplification was carried out as follows: after initial denaturation at 95°C for 5 min, 40 cycles of amplification at 95°C for 30 s, 63°C (methylated) or 57°C (unmethylated) for 30 s, and 72°C for 30 s, followed by a final extension of 72°C for 10 min. 10 μl of each PCR reaction were loaded and run onto 3% agarose gels stained with ethidium bromide and visualized under UV illumination. The PCR for all samples demonstrating methylation status of the CXCL12 gene was repeated to confirm these results.
Immunohistochemical analysis
Standard immunohistochemical detection was performed on sections from archival paraffin embedded breast tumor tissues and corresponding nonmalignant breast tissues. Protein expression in malignant and nonmalignant breast tissues were detected with specific antibodies against CXCL12 (anti-CXCL12, clone 79018, R&D systems, Minneapolis, MN; incubated for 2 h at 37°C), CXCR4 (anti-CXCR4, clone 44716, R&D systems, Minneapolis, MN; incubated for 2 h at 37°C), the estrogen receptor (ER) (Zymed, San Francisco, CA, USA; prediluted monoclonal mouse anti-human estrogen receptor clone 1D5, incubated for 18 h at 4°C), the progesterone receptor (PR) (Zymed, San Francisco, CA, USA; prediluted monoclonal mouse anti-human progesterone receptor clone PR-2C5, incubated for 18 h at 4°C), Her-2 (Zymed, San Francisco, CA, USA; prediluted monoclonal mouse anti-human Her-2 clone TAB250, incubated for 30 min at 37°C), p53 (Zymed, San Francisco, CA, USA; prediluted monoclonal mouse anti-human p53 protein clone BP53.12, incubated for 1 h at 37°C), and Ki-67 (Zymed, San Francisco, CA, USA; prediluted monoclonal mouse anti-human Ki-67 antigen clone 7B11, incubated for 1 h at 37°C). In addition, positive and negative controls for each marker were routinely performed during experiments.
Sections were then processed using the Histostain-SP kits according to the manufacturer’s recommendations (Zymed, San Francisco, CA, USA). Interpretation of the staining was carried out according to the usual criteria (Caldeira et al. 2006) by two independent blinded observers. If any discrepancies between classifications of samples arose, they were reviewed and the final results reached by consensus. In brief, scoring of CXCL12 and CXCR4 immunoreactivities were categorized into four semi-quantitative classes based on the rate of stained tumor cells: absence of staining, <10% positive cells (low), 10–50% positive cells (moderate), and >50% positive cells (high). We decided that the specimen should be regarded as CXCL12 or CXCR4 positive when the immunohistochemical staining was moderate or high, and negative when its staining was absent or low. ER, PR, and p53 were scored as follows: weak (1+), moderate (2+), and strong (3+) staining in >10% of the tumor cells or absent (0). Levels of Her-2 protein expression were evaluated semi-quantitatively using Zymed evaluation guidelines as follows: no staining or membrane staining in less than 10% of the tumor cells (0), weak, incomplete membrane staining in more than 10% of the tumor cells (1+), weak or moderate complete membrane staining in more than 10% of the tumor cells (2+), and strong, complete membrane staining in more than 10% of the tumor cells (3+). Further, for Ki-67 expression we evaluated the percentage of cells stained. The immunohistological markers were defined into dichotomous variables on the basis of the staining index such as negative expression levels (scores 0, 1, and 2), and positive staining (score 3) for ER, PR, Her-2 proteins. To p53 protein, the expression level was categorized as negative (scores 0 and 1) or positive (scores 2 and 3).
Statistical analysis
Descriptive statistics, such as mean, standard deviation, and percentage were used to summarize a patient’s data and gene expression or hypermethylation status. The correlations of the methylation alterations of the CXCL12 gene with clinicopathological parameters were analyzed by χ2 test. The distribution of categorical data between CXCL12 or CXCR4 immunostaining in breast cancers and clinicopathological characteristics were also assessed by χ2 test. The CXCL12 and CXCR4 mRNA expression levels among various subgroups were evaluated by using the Kruskal–Wallis test. The comparison of CXCL12, CXCR4 and DNMTs mRNA levels between different teams were calculated using the Mann–Whitney test. The Spearman rank correlation test was used to determine the links between continuous mRNA values of each DNMT. It was also used to determine the correlations between CXCL12 mRNA expression and CXCL12 methylation/protein expression, whereas correlation between CXCL12 methylation and CXCL12 protein expression was analyzed by χ2 test. Statistical significance was assumed for P < 0.05. The statistical tests were performed using the statistical software package SPSS, version 12.0 (LEAD Technologies, USA).
Results
Correlation of CXCL12 and CXCR4 mRNA levels with clinicopathological parameters in primary breast carcinoma
We first examined CXCL12 and CXCR4 mRNA expressions in all of the 63 primary breast carcinomas and the 20 normal breast tissues by semi-quantitative RT-PCR. A transcript of 145 bp corresponding to CXCR4 mRNA was detected in all tumor samples, whereas CXCR4 mRNA expression was only observed in 12 (60%) normal breast samples (Fig. 1a). The ligand CXCL12 mRNA amplification products were not observed in 18 (28.6%) breast carcinomas; the other 45 of 63 (71.4%) tumors expressed detectable levels of CXCL12 mRNA (Fig. 1b). All the 20 noncancerous tissues exhibited detectable CXCL12 mRNA products. To confirm whether a loss of CXCL12 mRNA expression actually existed, all analyses were repeated at least twice. GAPDH mRNA expression was detected in all samples.

Expression and methylation analyses of CXCL12/CXCR4 in primary breast carcinomas and normal breast tissues. a, b Representative expression of CXCR4 and CXCL12 mRNA in primary breast carcinomas (T) compared to the normal breast tissues (N) by semi-quantitative RT-PCR. c Methylation analysis of CXCL12 in normal breast tissues (N) and primary breast carcinomas (T) by methylation-specific PCR. Lane U unmethylated, Lane M methylated. Data in (a–c) are representative of two independent experiments
Correlation of real-time RT-PCR quantification of CXCR4 and CXCL12 genes with clinical data in the 63 breast carcinomas are shown in Tables 2, 3. The results showed that CXCR4 mRNA in primary breast carcinomas (1.22 ± 0.80, Mean ± SD) was over-expressed as compared to normal breast samples (0.65 ± 0.59, P = 0.001). CXCR4 over-expression in primary breast carcinomas was significantly related to lymph node metastasis (P = 0.013), and strong Her-2 expression (P = 0.031). In contrast to CXCR4, quantitative analysis of the CXCL12 transcript revealed significantly (P = 0.031) lower levels in breast carcinoma tissues (1.41 ± 1.18) than in normal mammary tissues (2.02 ± 0.71). Furthermore, significantly (P = 0.017) lower expression levels of CXCL12 were found in tumors with ≥3 positive lymph nodes metastasis. A significant difference (P = 0.047) in CXCL12 mRNA levels were also found between estrogen receptor negativity and positive group.
Immunohistochemical expression of CXCR4 and CXCL12 and their correlation with clinical–pathological data in primary breast carcinoma
In our series, the predominant staining pattern of CXCR4 or CXCL12 was cytoplasmic staining, which was in consistence with previous reports (Andre et al. 2006; Koshiba et al. 2000). Out of 63 breast carcinoma specimens tested, 50 (79.4%) stained positive for CXCR4. The positive cases were divided into two score classes according to their staining: moderate in 31 (62.0%) (Fig. 2b), and high in 19 (38.0%) (Fig. 2c). All normal breast specimens resulted in negative staining for CXCR4 (Fig. 2a). Table 3 summarizes the relationship between CXCR4 protein expression and clinicopathological features of the 63 breast cancers. χ2 analysis indicated a significant correlation (P = 0.035) between aberrant CXCR4 expression and lymph node metastasis, recorded as positive staining in all the 17 tumors (100%) with ≥3 positive lymph nodes metastasis compared with 33 of 46 tumors (71.7%) with <3 or no positive lymph nodes metastasis. Positive protein expression of CXCR4 appeared to be more frequent in Her-2 positive tumors (94.7%) than in Her-2 negative tumors (72.7%), although it did not reach the statistical significance (P = 0.101).

Immunohistochemical analysis of CXCR4 and CXCL12 protein in primary breast carcinomas and normal breast tissues. a A normal breast sample showing negative CXCR4 expression. b A breast tumor sample showing moderate CXCR4 expression. c A breast tumor sample showing high CXCR4 expression. d A normal breast sample showing positive CXCL12 expression. e A CXCL12-unmethylated breast tumor showing positive CXCL12 expression. f A CXCL12-methylated breast tumor showing negative CXCL12 expression. Panels a–f, ×400
All normal breast specimens exhibited positive staining for CXCR4 (Fig. 2d). Only 24 breast cancers (38.1%) showed CXCL12 staining of moderate to high intensity (Fig. 2e). The remaining 39 tumors (61.9%) showed a complete absent or low staining of CXCL12 protein (Fig. 2f). There was no significant correlation between the expression of CXCL12 protein and the clinicopathological variables examined (Table 2). In tumors with ≥3 positive lymph nodes metastasis, negative staining of CXCL12 was observed in 14 cases (82.4%), which tended to be high in comparison to groups with <3 or no positive lymph nodes metastasis, but the difference was not statistically significant (P = 0.120).
The methylation status of CXCL12 and their correlation with clinical–pathological data in primary breast carcinoma
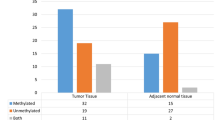
Methylation status of CXCL12 was detected in the 63 resected primary breast carcinomas of different clinical stages and in the corresponding nonmalignant breast samples (Fig. 1c). Methylation-specific PCR showed that 33 (52.4%) of 63 primary breast tumors were hypermethylated in the CXCL12 promoter region whereas, hypermethylation of CXCL12 was not observed in any of the 20 normal breast tissues. Among methylated tumors, we observed significant heterogeneity because both methylated and unmethylated products were present in 20 of 33 (60.6%) CXCL12-methylated tumors. The correlations of methylation frequencies with clinical, pathological, and biologic parameters were shown in Table 2. CXCL12 methylation was significantly associated with ≥3 positive lymph nodes metastasis (P = 0.032), and estrogen receptor negativity (P = 0.011). Of the 17 cases with ≥3 positive lymph nodes metastasis, 13 (76.5%) were methylated. Conversely, of the 34 cases with no or less than 3 positive lymph nodes metastasis, only 13 (38.2%) were methylated. CXCL12 methylation was also more frequently observed in tumors with estrogen receptor negativity with 72.0% (18/25) of the estrogen receptor negative cases identifies as CXCL12-methylation. None of the other factors including tumor grade, age, menopausal status, and histological type revealed a correlation with CXCL12 methylation.
Correlations between mRNA expression, protein expression, and DNA methylation in primary breast carcinoma
These 63 breast tumors were examined with all of the analyses, including protein, mRNA expression, and DNA methylation. These data were then cross-referenced to investigate the correlations between these three parameters. CXCR4 and CXCL12 protein expressions were significantly associated with their mRNA expressions (P = r = +0.511, 1.9 × 10−5; r = +0.534, 6.6 × 10−6, respectively).
Statistical analysis revealed that CXCL12 mRNA levels in primary breast tumors with hypermethylation (0.75 ± 0.71) were significantly (P = 4.6 × 10−7) lower than those without hypermethylation (2.12 ± 1.19). Of the 33 tumors exhibiting hypermethylation on the start site of the CXCL12 transcripton, 29 (87.9%) showed low expression (<1.69-fold), 2 (6.1%) showed similar expression (1.69–2.35), and 2 (6.1%) showed high expression levels of CXCL12 (>2.35-fold) compared to the mean of all the 20 normal breast samples (2.02-fold, with 95% confidence interval 1.69–2.35). Conversely, of the 30 tumors where the promoter region of CXCL12 was unmethylated, 10 (33.3%) showed no or low expression, 5 (16.7%) showed expression levels similar to the normal samples, and 15 (50.0%) had high CXCL12 expression levels compared to the mean of normal breast tissues. The Spearman rank correlation test revealed a significant inverse relationship between CXCL12 methylation status and their mRNA levels (r = −0.568, P = 1.2 × 10−6). A strong relationship between CXCL12 methylation and protein expression was also observed (P = 0.001).
DNMT enzymes are over-expressed in primary breast carcinoma and their association with clinical, pathological, and biological parameters in primary breast carcinoma
To define the mechanism leading to CXCL12 hypermethylation in primary breast carcinomas better, we determined and quantified the DNMTs expression status in these 63 breast cancers by semi-quantitative RT-PCR (Fig. 3a, b). In mammals, DNA methylation patterns are established and maintained by mainly three known functional DNMTs genes, namely DNMT1, 3A, and 3B, so we focused on these three genes. Furthermore, we sought links between CXCL12 methylation status and DNMTs mRNA expression levels from quantitative real-time PCR. Using the Spearman rank correlation test, we found that mRNA levels of the three DNMTs correlated strongly with each other, as follows: DNMT1 with DNMT3A (r = +0.272, P = 0.031), DNMT1 with DNMT3B (r = +0.303, P = 0.016), and DNMT3A with DNMT3B (r = +0.389, P = 0.002). Consistent with the hypothesis that over-expression of the enzymes catalyzing DNA methylation may be a prerequisite for abnormal DNA methylation (Robertson 2001), we determined that the expression levels of DNMT1 (1.57 ± 1.11, P = 0.012) and DNMT3B (1.66 ± 1.10, P = 0.026) were significantly higher in the CXCL12-methylated primary breast carcinomas than in the CXCL12-unmethylated ones (0.96 ± 0.69, 1.05 ± 0.99, respectively). But no significant difference (P = 0.185) of DNMT3A was observed between the CXCL12-methylated (1.63 ± 1.25) and CXCL12-unmethylated (1.22 ± 1.19) breast carcinomas. In addition, the levels of DNMT1, 3A, and 3B transcripts in the CXCL12-methylated astrocytomas were significantly higher than normal breast tissues (P = 0.008, 0.002, 1.4 × 10−5, respectively). Significant differences of DNMT3A (P = 0.025) and DNMT3B (P = 0.011) were also found between the CXCL12-unmethylated carcinomas and normal breast tissues. But no difference of DNMT1 (P = 0.475) was found between the CXCL12-unmethylated carcinomas and normal breast tissues (Fig. 3c).

Over-expressions of DNA methyltransferase enzymes in CXCL12-methylated breast carcinomas as compared with in CXCL12-unmethylated ones. a Representative mRNA expressions of DNMT1, DNMT3A, and DNMT3B in the CXCL12-methylated (Lanes 1–6) and CXCL12-unmethylated (Lanes 7–12) breast carcinomas by semi-quantitative RT-PCR. GAPDH served as endogenous control. b Semi-quantification of expression levels of DNMT1, 3A and 3B relative to GAPDH in these 12 breast carcinoma samples. c Quantitative real-time PCR analysis of gene expression levels of DNMT1, 3A and 3B in the CXCL12-methylated and CXCL12-unmethylated breast carcinoma samples, as well as normal breast tissues. Statistically significant differences are denoted by brackets with P values. In addition, the transcript levels of DNMT3A (P = 0.025) and DNMT3B (P = 0.011) in the CXCL12-unmethylated astrocytomas were significantly higher than normal breast tissues
Discussion
Formation of metastases requires several distinct steps, including intravasation from primary tumor into vascular or lymphatic circulation, resistance to apoptosis, trafficking to secondary organs, and proliferation of cancer cells in target organs and tissues (Chambers et al. 2002). CXCL12 binding to CXCR4 activates almost all of these key events (Luker and Luker 2006). Data has shown that over-expression of CXCR4 alone can significantly increase breast cancer metastasis to bone (Kang et al. 2003). In the present study, we found over-expression of both the transcript and protein levels of CXCR4 in primary breast carcinomas. Importantly, CXCR4 over-expression was significantly related to lymph node metastasis. Similar results were also observed by Kato et al. (2003). According to studies with cultured cells, it appears that CXCR4 is regulated by several genetic or biochemical agents at multiple levels including transcription, translation, and protein degradation (Luker and Luker 2006).
In blood-flow pattern of metastasis, mechanical factors determine the initial fate of cancer cells after leaving their primary tumor. The target organ is dictated by which the cells travel to first. Distinct from blood-flow pattern, some types of tumors show an organ-specific pattern of metastasis. There, both ‘seed’ (the cancer cell) and ‘soil’ (factors in the organ environment) contribute to this organ specificity (Chambers et al. 2002). As in the CXCL12/CXCR4 signaling axis, not only is the expression of CXCR4 a key determinant of tumor metastasis, but CXCL12 is also essential for site-specific metastatic process. CXCL12 exhibits peak levels of constitutive expression in organs representing the first destination of cancer metastasis (Müller et al. 2001). Nevertheless, contrary to the comprehensive assessment of CXCR4 expressions and regulatory mechanisms in primary tumors, studies defining CXCL12 expressions in various carcinomas are more limited. Previous studies have demonstrated that recombined human SDF-1 could stimulate the proliferation, migration, and invasion of a variety of tumor cells in vitro (Barbero et al. 2003; Sutton et al. 2007). Re-expression of CXCL12 increased mammary carcinoma cell proliferation and orthotopic primary mammary tumor growth (Wendt et al. 2008). Given the evidence demonstrating the pro-metastatic roles of CXCR4-CXCL12 signaling in tumor progression, one may have expected over-expression of CXCL12 in tumor cells to result in increased proliferation and metastasis (Kang et al. 2005; Koshiba et al. 2000; Scotton et al. 2002). However, our data have shown that primary breast carcinoma tissues had low levels of CXCL12 mRNA and protein expression as compared with normal mammary tissues. Furthermore, CXCL12 mRNA levels in ≥3 positive lymph nodes metastasis tumors were significantly lower than tumors with <3 or no positive lymph nodes metastasis. Similar results were also reported from colonic and mammary carcinoma cell lines (Wendt et al. 2006, 2008). CXCL12 transcript expression was undetectable in highly aggressive cells such as the MDA-MB-231 and MDA-MB-435s, whereas restricted expression of CXCL12 was only detected in the less aggressive carcinoma lines MCF-7 and MDA-MB-134 (Wendt et al. 2008). Also in gliomablastomas, CXCR4 was expressed in all nine tumor tissues, while CXCL12 is expressed in only one-third of tumors analyzed (Barbero et al. 2003). Herein, we propose that silencing of CXCL12 in metastatic cancer cells changes CXCL12-CXCR4 signaling from the autocrine and/or local paracrine loop to the endocrine loop. Metastatic cancer cells, lacking expression of CXCL12 but maintaining expression of CXCR4, pathologically follow the chemotactic signaling-endocrine CXCL12 gradients, enter the vascular or lymphatic circulation, survive in the vessels, actively invade ectopic tissues, and finally settle down in appropriate cellular niches. In light of this hypothesis, it is no surprise that re-expression of endogenous CXCL12 in colorectal or breast carcinoma cells dramatically reduced metastatic tumor formation in mice (Wendt et al. 2006, 2008).
Structural changes in the genome of tumors have been studied for decades of years and a series of candidate genes has been identified. Recently, there is increasing evidence that altered methylation patterns of tumor-associated genes are present in a variety of tumors, suggesting that DNA hypermethylation is also a major event in the stages of tumor progression (Waha et al. 2005). Previous studies have regarded CXCL12 as mainly regulated through degradation (Davis et al. 2005; Valenzuela-Fernández et al. 2002). However, recent evidence has identified DNA hypermethylation also involved in the silencing of CXCL12 in colonic and mammary cancer cells (Wendt et al. 2006, 2008). Herein, we focused on the correlation of promoter methylation of CXCL12 with clinical data in primary breast carcinomas. We detected the promoter regions of CXCL12 were hypermethylated as high as 52.4% in the 63 primary breast tumors, whereas hypermethylation of CXCL12 was not observed in any of the 20 normal breast tissues. Tumors with hypermethylated CXCL12 showed significantly lower expression levels of CXCL12, suggesting that hypermethylation down-regulates the transcription of this gene. More importantly, CXCL12 methylation was significantly associated with ≥3 positive lymph nodes metastasis. This might explain our observation that significantly lower expression levels of CXCL12 mRNA were found in cases with ≥3 positive lymph nodes metastasis.
As is known, physiologic DNA methylation is mainly achieved by three functional DNMT genes, namely DNMT1, 3A, and 3B. Over-expression of these enzymes is regarded as a prerequisite for abnormal DNA methylation (Robertson 2001). In the present study, over-expression of DNMT1 and DNMT3B mRNA was observed in the CXCL12-methylated group as compared with the CXCL12-unmethylated one. Previous reports have described DNMT1 mainly involved in maintaining the preexisting methylation pattern during replication, whereas DNMT3A and 3B mainly involved in de novo methylation (Bestor 2000). But as more studies reveal, there is a considerable level of cooperation and functional overlap among them (Siedlecki and Zielenkiewicz 2006). RNA interference-mediated knockdown of DNMT1 or DNMT3B induced CXCL12 expression in MCF-7 and AsPC1 cancer cells, while the demethylating agent 5-aza-dC exhibited the strongest effect on these cells (Sowińska and Jagodzinski 2007). In addition, previous study (Lapidus et al. 1998) has reported that over-expression of DNMTs can also inactivate the estrogen receptor gene. This might explain the correlation, found in this study, between CXCL12 hypermethylation and estrogen receptor negativity.
Conclusions
We have shown that epigenetic alteration plays an important role in the down-regulation of the expression levels of CXCL12 mRNA in breast cancers. Our results, together with recent findings emphasize the importance of the CXCL12/CXCR4 signaling axis in organ-specific pattern of metastasis. Cancer cells, lacking expression of CXCL12 but maintaining expression of CXCR4, pathologically follow the chemotactic signaling-endocrine CXCL12 gradients, shedding from a primary tumor, arresting in a particular new organ, and finally settling down in appropriate tissues. Both the ‘seed’ and the ‘soil’ contribute to this organ specificity.
Abbreviations
- CT :
-
Threshold cycle
- CXCR4:
-
CXC chemokine receptor-4
- CXCL12:
-
CXC chemokine ligand-12
- DNMT:
-
DNA methyltransferase
- ER:
-
Estrogen receptor
- GPCR:
-
G-protein coupled receptor
- MSP:
-
Methylation-specific polymerase chain reaction
- PR:
-
Progesterone receptor
- SDF-1:
-
Stromal cell-derived factor-1
References
Andre F, Cabioglu N, Assi H, Sabourin JC, Delaloge S, Sahin A et al (2006) Expression of chemokine receptors predicts the site of metastatic relapse in patients with axillary node positive primary breast cancer. Ann Oncol 17:945–951. doi:10.1093/annonc/mdl053
Arya M, Ahmed H, Silhi N, Williamson M, Patel HR (2007) Clinical importance and therapeutic implications of the pivotal CXCL12-CXCR4 (chemokine ligand-receptor) interaction in cancer cell migration. Tumour Biol 28:123–131. doi:10.1159/000102979
Barbero S, Bonavia R, Bajetto A, Porcile C, Pirani P, Ravetti JL et al (2003) Stromal cell-derived factor 1alpha stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1/2 and Akt. Cancer Res 63:1969–1974
Bestor TH (2000) The DNA methyltransferases of mammals. Hum Mol Genet 9:2395–2402. doi:10.1093/hmg/9.16.2395
Caldeira JR, Prando EC, Quevedo FC, Neto FA, Rainho CA, Rogatto SR (2006) CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer 6:48. doi:10.1186/1471-2407-6-48
Chambers AF, Groom AC, MacDonald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2:563–572. doi:10.1038/nrc865
Davis DA, Singer KE, De La Luz Sierra M, Narazaki M, Yang F, Fales HM et al (2005) Identification of carboxypeptidase N as an enzyme responsible for C-terminal cleavage of stromal cell-derived factor-1alpha in the circulation. Blood 105:4561–4568. doi:10.1182/blood-2004-12-4618
Girault I, Tozlu S, Lidereau R, Bièche I (2003) Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin Cancer Res 9:4415–4422
Grunau C, Clark SJ, Rosenthal A (2001) Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res 29:E65. doi:10.1093/nar/29.13.e65
Jiang Z, Li X, Hu J, Zhou W, Jiang Y, Li G et al (2006) Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci Res 56:450–458. doi:10.1016/j.neures.2006.09.006
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C et al (2003) A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3:537–549. doi:10.1016/S1535-6108(03)00132-6
Kang H, Watkins G, Parr C, Douglas-Jones A, Mansel RE, Jiang WG (2005) Stromal cell derived factor-1: its influence on invasiveness and migration of breast cancer cells in vitro, and its association with prognosis and survival in human breast cancer. Breast Cancer Res 7:R402–R410. doi:10.1186/bcr1022
Kato M, Kitayama J, Kazama S, Nagawa HBB (2003) Expression pattern of CXC chemokine receptor-4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res 5:R144–R150. doi:10.1186/bcr627
Kim J, Mori T, Chen SL, Amersi FF, Martinez SR, Kuo C et al (2006) Chemokine receptor CXCR4 expression in patients with melanoma and colorectal cancer liver metastases and the association with disease outcome. Ann Surg 244:113–120. doi:10.1097/01.sla.0000217690.65909.9c
Koshiba T, Hosotani R, Miyamoto Y, Ida J, Tsuji S, Nakajima S et al (2000) Expression of stromal cell-derived factor 1 and CXCR4 ligand receptor system in pancreatic cancer: a possible role for tumor progression. Clin Cancer Res 6:3530–3535
Kwon YM, Park JH, Kim H, Shim YM, Kim J, Han J et al (2007) Different susceptibility of increased DNMT1 expression by exposure to tobacco smoke according to histology in primary non-small cell lung cancer. J Cancer Res Clin Oncol 133:219–226. doi:10.1007/s00432-006-0160-2
Lapidus RG, Nass SJ, Davidson NE (1998) The loss of estrogen and progesterone receptor gene expression in human breast cancer. J Mammary Gland Biol Neoplasia 3:85–94. doi:10.1023/A:1018778403001
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. doi:10.1006/meth.2001.1262
Luker KE, Luker GD (2006) Functions of CXCL12 and CXCR4 in breast cancer. Cancer Lett 238:30–41. doi:10.1016/j.canlet.2005.06.021
Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME et al (2001) Involvement of chemokine receptors in breast cancer metastases. Nature 410:50–56. doi:10.1038/35065016
Robertson KD (2001) DNA methylation, methyltransferases, and cancer. Oncogene 20:3139–3155. doi:10.1038/sj.onc.1204341
Scotton CJ, Wilson JL, Scott K, Stamp G, Wilbanks GD, Fricker S et al (2002) Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res 62:5930–5938
Shim H, Lau SK, Devi S, Yoon Y, Cho HT, Liang Z (2006) Lower expression of CXCR4 in lymph node metastases than in primary breast cancers: potential regulation by ligand-dependent degradation and HIF-1alpha. Biochem Biophys Res Commun 346:252–258. doi:10.1016/j.bbrc.2006.05.110
Siedlecki P, Zielenkiewicz P (2006) Mammalian DNA methyltransferases. Acta Biochim Pol 53:245–256
Sowińska A, Jagodzinski PP (2007) RNA interference-mediated knockdown of DNMT1 and DNMT3B induces CXCL12 expression in MCF-7 breast cancer and AsPC1 pancreatic carcinoma cell lines. Cancer Lett 255:153–159. doi:10.1016/j.canlet.2007.04.004
Sutton A, Friand V, Brulé-Donneger S, Chaigneau T, Ziol M, Sainte-Catherine O et al (2007) Stromal cell-derived factor-1/chemokine (C-X-C motif) ligand 12 stimulates human hepatoma cell growth, migration, and invasion. Mol Cancer Res 5:21–33. doi:10.1158/1541-7786.MCR-06-0103
Valenzuela-Fernández A, Planchenault T, Baleux F, Staropoli I, Le-Barillec K, Leduc D et al (2002) Leukocyte elastase negatively regulates stromal cell-derived factor-1 (SDF-1)/CXCR4 binding and functions by amino-terminal processing of SDF-1 and CXCR4. J Biol Chem 277:15677–15689. doi:10.1074/jbc.M111388200
Waha A, Güntner S, Huang TH, Yan PS, Arslan B, Pietsch T et al (2005) Epigenetic silencing of the protocadherin family member PCDH-gamma-A11 in astrocytomas. Neoplasia 7:193–199. doi:10.1593/neo.04490
Wendt MK, Johanesen PA, Kang-Decker N, Binion DG, Shah V, Dwinell MB (2006) Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene 25:4986–4997. doi:10.1038/sj.onc.1209505
Wendt MK, Cooper AN, Dwinell MB (2008) Epigenetic silencing of CXCL12 increases the metastatic potential of mammary carcinoma cells. Oncogene 27:1461–1471. doi:10.1038/sj.onc.1210751
World Health Organisation (1981) International histological classification of tumours No. 2. Histological typing of breast tumours, 2nd edn. World Health Organisation, Geneva
Yang X, Yan L, Davidson NE (2001) DNA methylation in breast cancer. Endocr Relat Cancer 8:115–127. doi:10.1677/erc.0.0080115
Zlotnik A (2006) Chemokines and cancer. Int J Cancer 119:2026–2029. doi:10.1002/ijc.22024
Acknowledgments
We thank the staff of the Breast Cancer Center, Shandong Cancer Hospital and Institute for assistance in patient care and specimen collection. Written consent for publication was obtained from the patients or their relatives. Support was provided by the Natural Science Foundation of Shandong, 2006ZRC03115 and Y2005C39. This study was also supported by the National High Technology Research and Development Program of China (863 Program), 2007AA02Z437.
Conflict of interest statement
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Wei Zhou and Zheng Jiang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Zhou, W., Jiang, Z., Liu, N. et al. Down-regulation of CXCL12 mRNA expression by promoter hypermethylation and its association with metastatic progression in human breast carcinomas. J Cancer Res Clin Oncol 135, 91–102 (2009). https://doi.org/10.1007/s00432-008-0435-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-008-0435-x