
Abstract
There is no standard chemotherapy option for patients with biliary tract cancers. These patients present fairly ill and can have a rapid progression of disease. We conducted a multi-center, phase-II trial for patients with locally unresectable or metastatic bile duct or gallbladder adenocarcinomas using a modified regimen of gemcitabine and cisplatin to potentially improve tolerability. Patients received a 21-day treatment cycle of gemcitabine at 1,000 mg/m2 and cisplatin at 30 mg/m2 on days 1 and 8. To participate, 33 patients signed informed consent, and 30 patients received at least one dose of chemotherapy. By intention-to-treat analyses, 7 patients (21%) experienced a partial response and another 12 (36%) had stable disease for at least 12 weeks. The median progression-free survival was 6.3 months and median overall survival was 9.7 months. After 1 year, 39% of patients were alive. Most common grade 3–4 toxicities included neutropenia (33%), thrombocytopenia (23%), anemia (20%), nausea (20%), emesis (13%) and fatigue (10%). Of note, 52% of patients withdrew from study treatment, principally due to treatment-related adverse events. We concluded that this modified regimen appeared to have comparable activity to other gemcitabine and cisplatin regimens against advanced bile duct and gallbladder cancers, but there was still moderate toxicity in this patient population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cholangiocarcinomas and gallbladder cancers affect approximately 12,000 people in the United States annually [1]. Cholangiocarcinomas are classified by their location along the hepatobiliary system; this impacts resectability and survival. Bile duct cancers that are not surgically resectable have a rapid and fatal course. Similarly, tumors of the gallbladder are particularly aggressive when discovered by symptoms, rather than incidentally by cholecystectomy. The vast majority of cholangiocarcinomas and gallbladder cancers are adenocarcinomas and behave similarly when at an advanced stage.
Systemic control of biliary tract cancers has been challenging. There is no standard chemotherapy for these diseases. Biliary tract cancers are considered similar to pancreatic cancer in both aggressiveness and sensitivity to chemotherapy. After gemcitabine was determined to be active against advanced pancreatic cancer [2], it was tested in patients with metastatic biliary tract tumors. Response rates with single-agent gemcitabine have varied from 0% to 30%, with median overall survival (OS) ranging from 5 months to 14 months [3–11]. Combination regimens with gemcitabine have also generated interest [4, 12–24]. The goal of such combinations would be to improve upon efficacy while minimizing additional toxicities, since treatment for advanced biliary tract cancers is palliative.
Our group initiated a trial of gemcitabine and cisplatin in 2002 to determine the activity of the combination. Gemcitabine and cisplatin have a synergistic cytotoxic effect in vitro and in vivo [25, 26]. We chose a 21-day cycle treatment regimen with a relatively low dose of cisplatin to capitalize on this synergy and potentially reduce the likelihood of treatment-related toxicity.
Materials and methods
Patients
Patients were eligible for this study if they had locally advanced or metastatic bile duct or gallbladder adenocarcinoma and had undergone no more than one prior chemotherapy regimen. Patients were required to have measurable disease according to Response Evaluation Criteria in Solid Tumors Group (RECIST), had an Eastern Cooperative Oncology Group (ECOG) performance status 0–2, were at least 18 years old, had life expectancy of at least 12 weeks, and had adequate hematological (absolute neutrophil count at least 1,500/mm3 and platelets at least 100,000/mm3), hepatic (total bilirubin no more than 2.0 mg/dl; transaminases less than threefold upper limit of normal unless liver involved, in which case fivefold) and renal (creatinine no more than 1.8 mg/dl) function. Patients were excluded if they had: previously been treated with gemcitabine, prior chemotherapy within 3 weeks of initiation of treatment, a peripheral neuropathy of grade 2 or greater severity, major surgery in the past 4 weeks, uncontrolled serious medical or psychiatric illness, or other concurrent malignancy (except limited basal cell or squamous cell carcinoma of the skin or in situ cervix carcinoma). Patients were enrolled from the Dana-Farber Cancer Institute, Massachusetts General Hospital, and the Beth Israel Deaconess Medical Center, all in Boston, MA. The study was approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board. All patients signed informed consent.
Treatment
Treatment cycles were repeated every 21 days. Patients were treated on days 1 and 8 with intravenous gemcitabine 1,000 mg/m2 for a 30-min period, followed by cisplatin 30 mg/m2. All toxicities were graded according to the National Cancer Institute (NCI) Common Toxicity Criteria (CTC) version 2.0. Initiation of a new cycle required an absolute neutrophil count of at least 1,000/μl, platelets at least 100,000/μl, and resolution of other toxicities to be at least CTC grade 1. Day-8 therapy required an absolute neutrophil count of at least 1,000/μl and platelets at least 75,000/μl. Resolution of toxicity was required within 3 weeks of the intended start of the cycle, otherwise patients were withdrawn from the study.
Every week of treatment required both drugs to be administered; thus, if a toxicity was specifically attributed to cisplatin, resolution to protocol prescribed grade was required before either drug was readministered.
-For neutropenia, at 500–999 neutrophils/μl, or a platelet count less than 50,000–100,000/μl on day 1, chemotherapy was postponed until both counts exceeded these levels. If this took longer than 1 week, then gemcitabine and cisplatin were dose reduced to 75% of the prior dose.
-For neutropenia at less than 500/μl or a platelet count less than 50,000/μl on day 1, chemotherapy was postponed until absolute neutrophil count was at least 1,000/μl and platelets were at least 100,000/μl, at which time gemcitabine and cisplatin were restarted at 75% of prior dose.
-For neutropenia at 500–999/μl or platelet count less than 50,000–100,000/μl on day 8, gemcitabine and cisplatin were administered at 75% of prior dose, and this dose was continued for subsequent cycles.
-For neutropenia at less than 500/μl or platelet count less 50,000/μl on day 8, chemotherapy was postponed until absolute neutrophil count was at least 1,000/μl and platelets were at least 100,000/μl, at which time gemcitabine and cisplatin were restarted at 75% of prior dose and a new cycle was started.
Treatment was also interrupted if a patient’s creatinine measured greater than 1.8 mg/dl (or estimated creatinine clearance less than 50 ml/min). Treatment resumed at 75% of the initial dose of cisplatin (no change in gemcitabine) when these values returned to normal. Similarly, patients who developed peripheral neuropathy greater than grade 1 had treatment withheld, and it resumed at 75% of the prior cisplatin dose when neuropathy was no greater than grade 1. Treatment was also withheld from patients who developed other non-hematological toxicities (excluding alopecia, nausea, and vomiting) of a grade greater than 1; this resumed at 75% of the initial dose for both drugs when resolved to grade 1 or less.
Treatment was continued until development of progressive disease by RECIST, unacceptable toxicity, withdrawal of consent, intercurrent illness that prevented continuation of therapy, or changes in the patient’s condition that rendered him or her unable to continue study drugs (as judged by the treating clinician).
Evaluation
Baseline tumor measurements using computer tomography were obtained within 21 days before treatment was initiated. Physical examination, toxicity assessment, and laboratory studies were conducted on days 1 and 8 of each 3-week cycle, with the exception of the first two cycles when weekly assessments were required.
Repeat imaging was required at 6 and 12 weeks, and then every 9 weeks thereafter. Evaluation of response, stable disease, and disease progression was based on RECIST. Confirmation scans for responders (at least 30% reduction in the sum of the longest diameters of all measured lesions) were performed at least 4 weeks after the initial scan documenting the reduction.
Statistical analysis
The primary endpoint of this study was to determine the response rate of the combination of gemcitabine and cisplatin in patients with biliary tract or gallbladder adenocarcinoma. Secondary objectives included assessment of progression-free survival (PFS) and OS of the regimen as well as characterization of toxicities.
Responses were determined by RECIST, with an intention-to-treat analysis [27]. PFS was defined as the time between study enrollment and progression of disease or death while on protocol. OS was defined as the time between study enrollment and death. Median duration of response was defined as time between initial documentation of a partial response by RECIST criteria and progression of disease or death while on protocol. Patients who were withdrawn from the study for other reasons were censored at the discontinuation of study therapy.
Power calculations were based on a phase-II two-stage design. The proposed regimen was to be considered worthy for further investigation if a true response rate of 30% or greater was achieved and not worthy if it was 10% or less. A total of 30 eligible patients (defined as receiving at least one dose of therapy) were entered into the study in a two-stage design. Of these, 15 were entered in the first stage; once two responses were detected, an additional 15 patients entered the second stage. The probability of concluding the regimen effective after accruing 30 patients was 91% if the true response rate was 30 and 7% if it was 10%.
Results
Baseline characteristics
Between August 2002 and February 2005, 33 patients signed informed consent to participate in the trial at three medical centers in Boston, Massachusetts. Of these, 3 patients never started therapy due to medical complications prior to starting chemotherapy (n = 2) and withdrawal of consent prior to any therapy (n = 1). In addition, 4 patients came off study prior to the restaging scans being performed (due to toxicity, death, gastrointestinal bleed, and withdrawal of consent). The 30 patients who received at least one dose of therapy completed a median of 4 cycles (range 1–21+ cycles). Primary analyses were based on intent-to-treat; thus, all 33 patients who signed informed consent were included in efficacy analyses; only patients who received at least 1 dose of chemotherapy were included in toxicity analyses. The baseline characteristics of the enrolled patients are shown in Table 1. The study cohort was 60% male, with a median age of 57 years. Nearly 75% of the cohort had a baseline ECOG performance status of 1 or 2. No prior treatment had been given to 85% of the patients for their cancer. In total, 76% had intrahepatic cholangiocarcinomas, 15% gallbladder adenocarcinomas, and 9% extrahepatic cholangiocarcinomas.
Efficacy
The primary end point for this study was objective response rate (Table 2). By intent-to-treat analysis (n = 33), 7 patients (21%; 95% confidence interval [CI] 7–35%) experienced a partial response, and 12 patients (36%; 95% CI, 20–52%) had stable disease for at least 12 weeks. Among patients who completed at least two cycles of therapy (i.e., 6 weeks of treatment) and underwent restaging (excluding 3 patients who did not start protocol therapy and 4 patients who did not have any restaging), the response rate was 27% (95% CI, 10–44%) and incidence of stable disease for at least 12 weeks was 46% (95% CI, 33–59%). The median duration of response was 6.5 months (95% CI, 3.3–9.5 months). One patient with stable disease has remained on treatment for 14+ months, with minimal side effects.
Of the 33 patients, 52% withdrew from study treatment principally due to treatment-related adverse events—11 (33%) came off therapy due to toxicities requiring immediate withdrawal or that did not resolve within the required 3-week period and an additional 6 (19%) withdrew consent for further treatment due to treatment-related adverse events . Of the remainder, 10 patients (30%) came off therapy (Table 3) due to progression of disease, 1 withdrew consent so as to have a break from therapy after 8 months of treatment and another withdrew consent so as to pursue surgery. A further 3 patients never started treatment and, finally, 1 patient has remained in the study for the past 14 months.
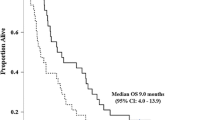
The median PFS for the entire patient cohort was 6.3 months (95% confidence interval [CI], 4.8–14.9 months). The median OS (Fig. 1) for the overall cohort was 9.7 months (95% CI, 6.4–13.8 months). At 1 year, 39% of patients were alive; at 18 months, 21% of patients were alive. PFS was 6.3 months and OS was 11.8 months when limited to the 26 patients that completed at least two cycles of protocol therapy (Fig. 2).

Progression-free survival for entire cohort (n = 33)

Overall survival for entire cohort (n = 33)
Toxicities
Patients received a median of 4 cycles of therapy (cycles = 21 days), with a range from 1 to 21+ cycles. Toxicities experienced by the patients treated with at least one dose of this regimen are shown in Table 4. Myelosuppression, gastrointestinal toxicities and fatigue were the most common side effects experienced. Grade 3 or 4 toxicities were modest, with 33% of patients with neutropenia, 23% with thrombocytopenia, 20% with anemia, 20% with nausea, 13% with vomiting and 10% with fatigue.
Of the patients in the treated cohort, 50% (n = 30) required a dose reduction of one or both agents while on trial. Of the 15 that did not require a dose reduction, 4 were on protocol therapy for less than 1 month, although 6 were on therapy for at least 4 cycles (3 or more months). The protocol permitted an unlimited number of 25% dose reductions of both gemcitabine and cisplatin (i.e., there was no lower limit of either drug requiring study withdrawal). A total of 14 (46%) patients required at least one dose reduction of the gemcitabine; 4 patients had one reduction, 7 had two, and 1 patient had each of three, four and five reductions. All but 2 patients had at least one of their gemcitabine reductions due to bone marrow suppression.
Similarly, a total of 15 patients (50%) had at least one dose reduction of cisplatin; 6 had one dose reduction, 6 had two and 1 had each of three, four, and five reductions. The majority of reductions were due to bone marrow suppression, although transient creatinine elevations as well as gastrointestinal toxicities accounted for eight of the overall number of reductions (representing 5 distinct patients).
Discussion
In this multi-institution, phase-II study of the combination of gemcitabine and cisplatin for patients with metastatic biliary tract cancers, we observed a response rate of 21% and median PFS of 6.3 months, by intent-to-treat. Of the patients, 57% had disease control for at least 12 weeks. The median survival for this population of patients was 9.7 months. At 1 year, 39% were alive. However, the regimen was still moderately toxic in this population (73% of whom had a performance status of 1 or 2), with 52% of patients coming off trial for toxicity-related issues and 70% having at least one grade 3 or 4 toxicity.
Biliary tract cancers are thought to act similarly to pancreatic cancers with regard to biological and clinical features; thus, many of the strategies to treat advanced pancreatic cancer have been tested in metastatic biliary tract cancer (Table 5). At present, gemcitabine seems to be the most active agent against this disease. Multiple phase-II trials of single-agent gemcitabine have shown remarkably varied results, with response rates ranging from 0% in a single trial of fixed dose rate infusion to 30% in two trials, and median OS values as short as 4.7 months to over 1 year. One potential explanation for this is the fact that gallbladder cancers and cholangiocarcinomas are often mixed in the same trial (only a few trials isolate one relative to the other) and may be more different than assumed. Alternatively, the etiology of these diseases may differ by region of the world and thus may differ biologically in their sensitivity to chemotherapies. Nonetheless, gemcitabine seems to be consistently active in a portion of patients with bile tract cancers.
We initiated this current trial during a period of active research of gemcitabine combinations in treatment of both pancreatic cancer and biliary tract cancers. Though most randomized phase-III trials of gemcitabine combinations for pancreatic cancer have not definitively shown a survival advantage when compared with gemcitabine alone [28], a pooled analysis of two randomized trials of gemcitabine with platinum analogs showed a 34% improvement (P = 0.003) in PFS and 23% improvement (P = 0.03) in OS with combination therapy relative to gemcitabine alone [29].
Four studies of gemcitabine and cisplatin and one study of gemcitabine and oxaliplatin for biliary tract cancers have been published. Response rates have ranged from 24% to 53%, median PFS has ranged from 3 months to 8 months and median OS from 4.6 months to 15.4 months. The current study lies within these ranges and confirms the general consensus of most of the studies that the true PFS is likely to be 4–6 months and OS under 1 year for patients with advanced biliary tract cancers. We utilized an intent-to-treat analyses; as some reflection of the severity of this malignancy, 3 patients wanting to join the trial had to be withdrawn prior to any treatment and 4 did not complete two cycles of therapy. We believe this drop-out rate mimics the real-world experience that oncologists face when considering therapy for patients with biliary tract cancers. Furthermore, the activity did not meet our initial hypothesis that a 30% observed response rate warrants further investigation.
The additional toxicities of even a low dose of cisplatin in this multi-center American population should be considered. Of the patients studied, 30% had grade 1 or 2 vomiting and 13% had grade 3. Bone marrow suppression was fairly prominent, requiring dose reductions in one-half of patients.
In conclusion, in this multi-center study, we demonstrated activity of gemcitabine and cisplatin in patients with metastatic biliary tract cancers but also moderate toxicity requiring dose reductions and study withdrawal in a number of patients. Our original hypothesis of a 30% response rate was not met. Nonetheless, there is an ongoing European randomized phase-III trial in patients with advanced biliary tract cancers that will ultimately determine whether the addition of cisplatin to gemcitabine treatments can improve survival beyond that observed with gemcitabine alone [24].
References
Jemal A, Siegel R, Ward E et al (2006) Cancer statistics, 2006. CA Cancer J Clin 56:106–130
Burris HA 3rd, Moore MJ, Andersen J et al (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15:2403–2413
Eng C, Ramanathan RK, Wong MK et al (2004) A phase II trial of fixed dose rate gemcitabine in patients with advanced biliary tree carcinoma. Am J Clin Oncol 27:565–569
Gebbia V, Giuliani F, Maiello E et al (2001) Treatment of inoperable and/or metastatic biliary tree carcinomas with single-agent gemcitabine or in combination with levofolinic acid and infusional fluorouracil: results of a multicenter phase II study. J Clin Oncol 19:4089–4091
Kubicka S, Rudolph KL, Tietze MK et al (2001) Phase II study of systemic gemcitabine chemotherapy for advanced unresectable hepatobiliary carcinomas. Hepatogastroenterology 48:783–789
Lin MH, Chen JS, Chen HH et al (2003) A phase II trial of gemcitabine in the treatment of advanced bile duct and periampullary carcinomas. Chemotherapy 49:154–158
Okusaka T, Ishii H, Funakoshi A et al (2006) Phase II study of single-agent gemcitabine in patients with advanced biliary tract cancer. Cancer Chemother Pharmacol 57:647–653
Park JS, Oh SY, Kim SH et al (2005) Single-agent gemcitabine in the treatment of advanced biliary tract cancers: a phase II study. Jpn J Clin Oncol 35:68–73
Penz M, Kornek GV, Raderer M et al (2001) Phase II trial of two-weekly gemcitabine in patients with advanced biliary tract cancer. Ann Oncol 12:183–186
Tsavaris N, Kosmas C, Gouveris P et al (2004) Weekly gemcitabine for the treatment of biliary tract and gallbladder cancer. Invest New Drugs 22:193–198
von Delius S, Lersch C, Schulte-Frohlinde E et al (2005) Phase II trial of weekly 24-hour infusion of gemcitabine in patients with advanced gallbladder and biliary tract carcinoma. BMC Cancer 5:61
Alberts SR, Al-Khatib H, Mahoney MR et al (2005) Gemcitabine, 5-fluorouracil, and leucovorin in advanced biliary tract and gallbladder carcinoma: a North Central Cancer Treatment Group phase II trial. Cancer 103:111–118
Andre T, Tournigand C, Rosmorduc O et al (2004) Gemcitabine combined with oxaliplatin (GEMOX) in advanced biliary tract adenocarcinoma: a GERCOR study. Ann Oncol 15:1339–1343
Bhargava P, Jani CR, Savarese DM et al (2003) Gemcitabine and irinotecan in locally advanced or metastatic biliary cancer: preliminary report. Oncology (Williston Park) 17:23–26
Cho JY, Paik YH, Chang YS et al (2005) Capecitabine combined with gemcitabine (CapGem) as first-line treatment in patients with advanced/metastatic biliary tract carcinoma. Cancer 104:2753–2758
Doval DC, Sekhon JS, Gupta SK et al (2004) A phase II study of gemcitabine and cisplatin in chemotherapy-naive, unresectable gall bladder cancer. Br J Cancer 90:1516–1520
Hsu C, Shen YC, Yang CH et al (2004) Weekly gemcitabine plus 24-h infusion of high-dose 5-fluorouracil/leucovorin for locally advanced or metastatic carcinoma of the biliary tract. Br J Cancer 90:1715–1719
Kim ST, Park JO, Lee J et al (2006) A phase II study of gemcitabine and cisplatin in advanced biliary tract cancer. Cancer 106:1339–1346
Knox JJ, Hedley D, Oza A et al (2005) Combining gemcitabine and capecitabine in patients with advanced biliary cancer: a phase II trial. J Clin Oncol 23:2332–2338
Knox JJ, Hedley D, Oza A et al (2004) Gemcitabine concurrent with continuous infusional 5-fluorouracil in advanced biliary cancers: a review of the Princess Margaret Hospital experience. Ann Oncol 15:770–774
Kornek GV, Schuell B, Laengle F et al (2004) Mitomycin C in combination with capecitabine or biweekly high-dose gemcitabine in patients with advanced biliary tract cancer: a randomised phase II trial. Ann Oncol 15:478–483
Kuhn R, Hribaschek A, Eichelmann K et al (2002) Outpatient therapy with gemcitabine and docetaxel for gallbladder, biliary, and cholangio-carcinomas. Invest New Drugs 20:351–356
Thongprasert S, Napapan S, Charoentum C et al (2005) Phase II study of gemcitabine and cisplatin as first-line chemotherapy in inoperable biliary tract carcinoma. Ann Oncol 16:279–281
Valle W, Wasan H, Johnson P et al (2006) Gemcitabine, alone or in combination with cisplatin, in patients with advanced or metastatic cholangiocarcinoma (CC) and other biliary tract tumors: a multicenter, randomized, phase II (the UK ABC-01) study, Gastrointestinal Cancer Symposium, pp Abstr 98
Bergman AM, Ruiz van Haperen VW, Veerman G et al (1996) Synergistic interaction between cisplatin and gemcitabine in vitro. Clin Cancer Res 2:521–530
Peters GJ, Bergman AM, Ruiz van Haperen VW et al (1995) Interaction between cisplatin and gemcitabine in vitro and in vivo. Semin Oncol 22:72–79
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Rocha Lima CM, Flores AM (2006) Gemcitabine doublets in advanced pancreatic cancer: should we move on? J Clin Oncol 24:327–329
Heinemann V, Labianca R, Hinke A et al (2006) Superiority of gemcitabine plus platinum analog compared to gemcitabine alone in advanced pancreatic cancer: pooled analysis of two randomized trials, the GERCOR/GISCAD Intergroup Study and a German Multicenter Study, Gastrointestinal Cancer Symposium, pp Abstr 96
Acknowledgment
Supported by Lilly Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Meyerhardt, J.A., Zhu, A.X., Stuart, K. et al. Phase-II Study of Gemcitabine and Cisplatin in Patients with Metastatic Biliary and Gallbladder Cancer. Dig Dis Sci 53, 564–570 (2008). https://doi.org/10.1007/s10620-007-9885-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-007-9885-2