
Abstract
This study was performed to evaluate whether down-regulation of prostaglandin E2 (PGE2) synthesis by celecoxib treatment is associated with inhibition of cell growth in human colon carcinoma cell lines. Physiologic concentrations of celecoxib (5–10 μM) inhibited 80% to 90% of PGE2 production in HT-29 cells that express high levels of COX-2 protein. In these concentrations, celecoxib had a minor inhibitory effect (20–30%) on cell growth. There was a significant change in induction of apoptosis only at higher concentrations of celecoxib (>20μM). Treatment by low concentrations of celecoxib did not alter the levels of COX-1, β-catenin, P27, Bcl-2, and Bcl-x proteins. The effect of celecoxib on cell growth inhibition was higher on the COX-2-positive HT-29 cell line (IC50=20μM) than on the COX-2 deficient SW-480 cell line (IC50=35μM). In conclusion, inhibition of PGE2 synthesis is an early, but not sufficient, step in the mechanism of celecoxib-mediated cell growth inhibition. These results support the need for additional evaluation of independent COX-2 pathways of celecoxib in chemoprevention of CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Colorectal cancer (CRC) is one of the leading causes of cancer deaths among both men and women in the western world [1]. Several lines of evidence suggest that cyclooxygenase-2 (COX-2) is one of the key factors in carcinogenesis. There are three isoforms of the COX enzyme. COX-1 is found in the normal gastrointestinal mucosa and is usually constitutively expressed. It serves as the housekeeping protein. The COX-2 gene was discovered a decade ago [2]. It is usually undetectable in the normal gastrointestinal mucosa, but its expression can be induced by inflammatory and neoplastic stimuli [3, 4]. Several studies have shown that COX-2 mRNA and protein levels are up-regulated in transformed cells [5, 6] as well as in both premalignant and malignant tissues, such as colorectal, pancreatic, and gastric adenocarcinomas [7]. Up-regulation of COX-2 expression occurs in 40% to 50% of colorectal polyps and in up to 85% of CRC [8].
The lack of COX-2 expression in the normal colonic mucosa together with its increased expression in colonic neoplasm constitute the rationale for the selective action of COX-2 inhibitors on neoplastic colonic mucosa, without major biologic effects on the normal colonic mucosa [9]. Recent studies have shown that treatment with selective COX-2 inhibitors caused cell growth inhibition and significant tumor suppression in the initiation as well as the promotion and progression phases of azoxymethane-induced colon cancer [10, 11]. Their antineoplastic properties have been primarily attributed to their ability to block COX-2 activity, which results in the inhibition of proliferation and the induction of apoptosis [12, 13]. Several in vitro, in vivo, and clinical studies have previously indicated that celecoxib, a specific COX-2 inhibitor, may prevent CRC [14–17]. Specifically, this drug significantly reduced the occurrence of tumor burden in animal models of intestinal neoplasia [14], it inhibited the growth of adenomatous polyps in patients with familial adenomatous polyposis [15] and it reduced the development of colorectal neoplasia in a general population of patients aged 65 years and older [16]. In an open-labeled study, we demonstrated that rofecoxib at 25 mg per day for up to 30 months prevented the growth of 80% of adenomas in patients with familial adenomatous polyposis [17]. Unfortunately, the issue of long-term safety with COX-2 inhibitors was recently questioned because of the increased cardiovascular and thrombotic toxicity that had been observed with their long-term use [18].
The antineoplastic properties of celecoxib have been attributed, at least in part, to its ability to directly inhibit COX-2 by binding to its active site [19]. Other studies, however, suggest that celecoxib may reduce the formation of polyps by COX-2-independent mechanisms, such as inhibition of the activation of Akt [20] or suppression of the ability of the PPARδ-receptor complex to bind to DNA [21].
This study was designed to evaluate whether down-regulation of PGE2 by celecoxib treatment is associated with the inhibition of cell growth or the induction of apoptosis in human colon carcinoma cell lines.
Materials and methods
Reagents and chemicals
Celecoxib was provided by Pfizer (New York, NY). Antibodies to COX-1, β–catenin, P21, P27, Bcl-2, and Bcl-x were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other reagents were of the highest purity and purchased from Sigma Chemical Co. (St. Louis, MO).
Cell culture
The growth inhibition of celecoxib was tested in human colon carcinoma cell lines HT-29 and SW480, which were obtained from the American Type Culture Collection (ATCC). The different cell lines were grown and maintained in DMEM (Biological Industries, Israel) supplemented with 5% fetal calf serum (FCS), 1% penicillin, and 1% streptomycin at 37°C, in an atmosphere of 95% oxygen and 5% CO2 (full medium).
Assays for inhibition of cell growth
The cells were plated in duplicate at a density of 3×104 in 12-well plates containing 1 ml of full medium as previously described [22]. Celecoxib or 0.1% dimethyl sulfoxide (DMSO, the drug vehicle) were added at the selected concentrations to the culture medium 24 hr after plating. The numbers of viable cells were determined in duplicate using a Coulter counter after 72 hr. All experiments were repeated at least three times and yielded similar results.
Flow-cytometric analysis
HT-29 cells were plated at a density of 5×106 per 10-cm dish with test drugs at selected concentrations. The adherent and nonadherent cells were collected during exponential growth of the cells and counted. A total of 1 to 2×106 cells were washed in phosphate-buffered saline (PBS) and the pellet was fixed in 3 ml of ethanol for 1 hr at 4°C. The cells were pelleted and resuspended in 1 ml of PBS and then incubated for 30 min with 0.64 mg/ml RNAse at 37°C. They were stained with 45 μg/ml of propidium iodide (PI) for at least 1 hr before analysis by flow cytometry using a standard protocol for cell cycle distribution and cell size [23]. Necrotic cells were counted using trypan blue before fixation. All experiments were performed three times and yielded similar results. Data acquisition was performed on a FACScan and analyzed using CellQuest software (Becton Dickinson Immunocytometry Systems, San Jose, CA). All fluorescence and laser light-scatter measurements were made with linear signal processing electronics. Data for at least 10,000 cells were collected for each data file.
Fluorescence microscopy
HT-29 cells were plated at a density of 5×106 per 10-cm dish with celecoxib at selected concentrations for 72 hr. Apoptotic HT-29 cells were detected by nuclear morphologic changes using PI staining. The cells were washed twice with PBS and fixed for 15 min at room temperature with 4% paraformaldehyde in PBS. The fixative was removed by aspiration, and the monolayer was washed twice in PBS. DNA was incubated with 0.15 mg/ml RNAse for 15 min and stained with 5 μg/ml PI at room temperature. Excess PI stain was removed, and the monolayer was thoroughly washed with PBS. The coverslip was mounted with glycerol and the stained nuclei were viewed at ×63 using a Leica TCS SP2 confocal microscope (Leica Microsystems, Wetzler, Germany).

Celecoxib at physiologic levels inhibits COX-2 activity but not its protein or mRNA levels. HT-29 cells were treated and incubated with celecoxib for 72 hr at selected doses. (A) PGE2 levels in the culture medium were measured by enzyme immunoassay (see Materials and methods). The data are mean values ± SD from three independent experiments. (B) The cells were collected for Western blot analysis (see Materials and methods). Upper panel: COX-2 expression; Lower panel: actin expression. The data are representative of three individual experiments. (C) The mRNA was extracted and equal amounts were transcribed to cDNA from a kit. The cDNA was taken from samples at various times and used as the DNA templates for polymerase chain reaction. Primers for COX-2 were loaded and visualized (see Materials and methods). GAPDH was used to ensure equal loading. The data are representative of three individual experiments
Protein extraction and Western blotting
Exponentially growing cells were collected and washed three times in ice-cold PBS. The cell pellets were resuspended in lysis buffer (20 mM Tris-HCI pH 7.4, 2 mM EDTA, 6 mM 6-mercaptoethanol, 1% NP-40, 0.1% SDS, and 10 mM NaF, plus the protease inhibitors leupeptin 10 μg/ml, aprotinin 10 μg/ml, and 0.1 mM phenylmethylsulfonylfluoride). The protein concentration of each sample was estimated using the Bio-Rad protein assay (Bio-Rad Laboratories, CA). For Western blotting, samples containing 50 μg total cell lysate were loaded onto a 10% SDS polyacrylamide gel and subjected to electrophoresis. Proteins were transferred to “Hybond-C” membranes (Amersham, Arlington Heights, IL) in transfer buffer (25 mM Tris, 190 mM glycine, 20% methanol), using a Trans Blot transfer apparatus at 70 mA for 12 to 18 hr at room temperature. The membranes were blocked with blocking buffer (PBS / 0.2% Tween 20 / 0.5% gelatin) for 1 hr at room temperature and subsequently washed three times for 5 min in washing buffer (PBS / 0.05% Tween-20). The membranes were incubated with monoclonal human anti-COX–1, anti-COX–2, anti-β-catenin, anti-Bcl-X, anti-Bcl-2, and anti-P27 antibodies for 1 hr at room temperature. The membranes were washed as described above and incubated with anti-goat (actin, COX-1, COX-2) or anti-rabbit (P-27, β–catenin, Bcl-X and Bcl-2) secondary antibodies (1:2000, 1:2000, and 1:10000, respectively) for 1 hr at room temperature. Additional washes were performed as described above, and immune detection was performed using the ECL Western blotting detection system (Amersham).
Measurement of PGE2 levels
The PGE2 concentration released by the cells was determined by a PGE2-specific enzyme-linked immunoassay (R&D Biosystems, Abingdon, UK) according to the manufacturer’s protocol.
Reverse transcriptase polymerase chain reaction (PCR)
HT-29 cells were treated with celecoxib for 72 hr as indicated. The mRNA was extracted, and equal amounts were transcribed to cDNA from a kit (Promega, USA). The cDNA was taken from samples at various times and used as the DNA templates for PCR. Primers used for COX-2 were 5′-TTC AAA TGA GAT TGT GGG AAA AT-3′ and 5′-AGA TCA TCT CTG CCT GAG TAT CTT-3′. GAPDH was used to ensure equal loading. Two milliliters was used as the template and 1 ml of each primer was used for each cDNA sample. The samples went through 30 rounds of PCR. They were separated on 1% agarose gel and visualized by ethidium bromide. The difference in RNA expression was analyzed by densitometric scanning.
Statistical analysis
The results were measured as mean ± SD. The difference between the intact and treated cells was evaluated by the one-way Student t-test using an SPSS(R) software package (SPSS, Inc., Chicago, IL). Statistical significance (P < 0.05) was established by the post hoc Tukey’s pairwise comparison.
Results
Physiological levels of celecoxib inhibits COX-2 activity
PGE2 synthesis was evaluated in HT-29 cells as a measure of COX-2 activity. Treatment with low concentrations of celecoxib (5–10 μM) resulted in 80% to 90% inhibition of PGE2 production (Fig. 1A). Western blot analysis showed that celecoxib did not affect COX-2 protein levels (Fig. 1B). Similarly, RT-PCR assay demonstrated that the level of COX-2 mRNA was not altered by treatment with celecoxib at physiologic levels (Fig. 1C).
Effect of celecoxib on levels of COX-1, β-catenin, P27, Bcl-2, and Bcl-X proteins
Western blot analysis showed that the level of COX-1 was not altered by treatment of celecoxib (Fig. 2). Similarly, treatment by low concentrations of celecoxib did not alter the levels of β-catenin, P27, Bcl-2, and Bcl-x proteins.

Celecoxib does not alter the levels of Cox-1, β-catenin, P27, Bcl-2, and Bcl-X proteins. HT-29 cells were incubated with 10 μM celecoxib for 72 hr and then collected and studied as described in Materials and methods. The treatment did not alter expression of COX-1, β-catenin P27, Bcl-2, and Bcl-x proteins. Lane 1, untreated HT-29 cells (control); lane 2, HT-29 cells treated with 10 μmol/L celecoxib; Lower panel: actin expression
Effect of physiologic levels of celecoxib on cell growth of human colorectal cancer cells
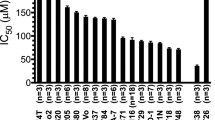
An inhibitory effect celecoxib on cell growth was found to be dose-dependent in both the HT-29 and SW-480 cell lines. Physiologic levels of celecoxib (5 μM) had only a minor inhibitory effect (20%) on the growth of the HT-29 cells that express high levels of COX-2 protein and no effect on the growth of the SW-480 cells that do not express COX-2 (Fig. 3) [24]. The effect of celecoxib on cell growth inhibition was significantly higher on the HT-29 cell line (IC50 = 20 μM) than on the SW-480 cell line (IC50 = 35 μM).
Effect of celecoxib on induction of apoptosis
Celecoxib increased the percentage of cells with sub-diploid DNA content, the hallmark of apoptosis in both the HT-29 and SW-480 cell lines (Fig. 4) only in high concentrations (>20 μM). Drug-treated HT-29 cells were examined by fluorescence microscopy for morphologic characteristics that would provide evidence of apoptosis. Typical apoptotic features of chromatin condensation and nuclear fragmentation confirmed the results of the FACS analysis (data not shown).

Effect of celecoxib on cell growth of human colorectal cancer. HT-29 and SW-480 cell lines were exposed to different concentrations of celecoxib for 72 hr as indicated. The data are mean ± SD values from three individual experiments performed in duplicate

Effect of celecoxib on induction of apoptosis. HT-29 and SW-480 cells were treated and incubated with celecoxib and harvested for quantification of apoptosis by flow cytometry (see MATERIALS AND METHODS). Data are mean ± SD values from three individual experiments performed in duplicate
Discussion
In the current study, we show that down-regulation of PGE2 synthesis by celecoxib is an early, but not sufficient, step in the mechanism of celecoxib-mediated cell growth inhibition or induction of apoptosis in CRC cell lines.
Several studies have shown that the serum physiologic concentration of celecoxib found to inhibit CRC tumor growth of in vivo models [28, 29] and human clinical trials [16, 30] is ∼2 to 5 μM. The precise mechanisms by which celecoxib inhibits proliferation of colon tumor cells are not yet fully understood. At this low concentration, the main biochemical target of celecoxib is thought to be the COX-2 isoenzyme [31–33]. Indeed, our results demonstrated that low concentrations of celecoxib (i.e., 5 μM) inhibited PGE2 production by >80%. This effect did not involve down-regulation of COX-2 mRNA or protein levels. Our finding is compatible with those of other studies in showing that celecoxib directly inhibits COX-2 by binding to its active site [20].
Our data show that COX-2 inhibition was associated with only a minor effect on cell growth. The concentration that was required to induce significant growth inhibition (>50%) was more than fourfold (20 μM) higher than levels required to inhibit COX-2 activity. As such, it is reasonable to conclude that the growth inhibition effect probably involves other pathways independent of PGE2 production.
Programmed cell death, apoptosis, is another putative target for nonsteroidal anti-inflammatory drugs (NSAIDs) and COXIBs [34, 35]. We had previously shown that celecoxib induced G2/M arrest, followed by induction of apoptosis in the transformed but not in the normal cells [36]. We now demonstrated that whereas celecoxib at physiologic levels inhibits COX-2 activity, it does not affect the induction of apoptosis. Specifically, apoptosis was induced by celecoxib only at higher concentrations, i.e., >20 μM, which is fourfold higher than maximal physiologic levels.
Recent studies identified a series of non-COX-2 molecular targets and signaling pathways that are involved in the effect of COXIBs and NSAIDs on cell growth inhibition and the induction of apoptosis. These targets include P27 [37], β-catenin [38], bcl-X, and bcl-2 [39]. Our results show that celecoxib at physiologic levels does not alter the level of any of these proteins, suggesting that other targets are involved. Additional experiments will be required to address the precise underlying mechanism.
In conclusion, inhibition of COX-2 enzyme is an early, but not sufficient, step in the mechanism of celecoxib-mediated cell growth inhibition and in the induction of apoptosis in CRC cell lines. Other independent COX-2 pathways are probably associated with the chemopreventive properties of celecoxib in vitro.
References
Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ (2005) Cancer statistics 2005. CA Cancer J Clin 55:10–30
Hla T, Neilson K (1992) Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A 89:7384–7388
Dannenberg AJ, Altorki NK, Boyle JO, Dang C, Howe LR, Weksler BB, Subbaramaiah K (2001) Cyclo-oxygenase 2: a pharmacological target for the prevention of cancer. Lancet Oncol 2:544–551
Tsujii M, DuBois RN (1997) Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 84:493–501
Subbaramaiah K, Telang N, Ramonetti JT, Araki R, DeVito B, Weksler BB, Dannenberg AJ (1996) Transcription of cyclooxygenase-2 is enhanced in transformed mammary epithelial cells. Cancer Res 56:4424–4429
Sheng GG, Shao J, Sheng H, Hooton EB, Isakson PC, Morrow JD, Coffey RJ Jr, DuBois RN, Beauchamp RD (1997) A selective cyclooxygenase-2 inhibitor suppresses the growth of H-ras-transformed rat intestinal epithelial cells. Gastroenterology 113:1883–1891
Koki A, Khan NK, Woerner BM, Dannenberg AJ, Olson L, Seibert K, Edwards D, Hardy M, Isakson P, Masferrer JL (2002) Cyclooxygenase-2 in human pathological disease. Adv Exp Med Biol 507:177–184
Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN (1994) Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 107:1183–1188
Chan TA (2002) Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol 3:166–174
Reddy BS, Rao CV, Seibert K (1996) Evaluation of cyclooxygenase-2 inhibitor for potential chemopreventive properties in colon carcinogenesis. Cancer Res 56:4566–4569
Fukutake M, Nakatsugi S, Isoi T, Takahashi M, Ohta T, Mamiya S, Taniguchi Y, Sato H, Fukuda K, Sugimura T, Wakabayshi K (1998) Suppressive effects of nimesulide, a selective inhibitor of cyclooxygenase-2, on azoxymethane-induced colon carcinogenesis in mice. Carcinogenesis 19:1939–1942
Dempke W, Rie C, Grothey A, Schmoll HJ (2001) Cyclooxygenase-2: a novel target for cancer chemotherapy? J Cancer Res Clin Oncol 127:411–417
Prescott SM, Fitzpatrick FA (2000) Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta 1470:69–78
Kawamori T, Rao CV, Seibert K, Reddy BS (1998) Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res 58:409–412
Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T, Su LK, Levin B (2000) The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 342:1946–1952
Rahme E, Barkun AN, Toubouti Y, Bardou M (2003) The cyclooxygenase-2-selective inhibitors rofecoxib and celecoxib prevent colorectal neoplasia occurrence and recurrence. Gastroenterology 125:404–412
Hallak A, Alon-Baron L, Shamir R, Moshkowitz M, Bulvik B, Brazowski E, Halpern Z, Arber N (2003) Rofecoxib reduces polyp recurrence in familial polyposis. Dig Dis Sci 48:1998–2002
Drazen JM (2005) COX-2 inhibitors: a lesson in unexpected problems. N Engl J Med 352:1131–1132
Hood WF, Gierse JK, Isakson PC, Kiefer JR, Kurumbail RG, Seibert K, Monahan JB (2003) Characterization of celecoxib and valdecoxib binding to cyclooxygenase. Mol Pharmacol 63:870–877
Kulp SK, Yang YT, Hung CC, Chen KF, Lai JP, Tseng PH, Fowble JW, Ward PJ, Chen CS (2004) Phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res 64:1444–1451
Wu GD (2000) A nuclear receptor to prevent colon cancer. N Engl J Med 342:651–653
Lev-Ari S, Strier L, Kazanov D, Madar-Shapiro L, Dvory-Sobol H, Pinchuk I, Marian B, Lichtenberg D, Arber N (2005) Celecoxib and curcumin synergistically inhibit the growth of colorectal cancer cells. Clin Cancer Res 11:6738–6744
Arber N, Han EK, Sgambato A, Piazza GA, Delohery TM, Begemann M, Weghorst CM, Kim NH, Pamukcu R, Ahnen DJ, Reed JC, Weinstein IB, Holt PR (1997) A K-ras oncogene increases resistance to sulindac-induced apoptosis in rat enterocytes. Gastroenterology 113:1892–1900
Smith ML, Hawcroft G, Hull MA (2000) The effect of nonsteroidal anti-inflammatory drugs on human colorectal cancer cells: evidence of different mechanisms of action. Eur J Cancer 36:664–674
Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA (2000) The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the Min mouse model of adenomatous polyposis. Cancer Res 60:5040–5044
Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, Paulson S, Seibert K, Rao CV (2000) Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res 60:293–297
Gupta RA, Dubois RN (2001) Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 1:11–21
Williams CS, Watson AJ, Sheng H, Helou R, Shao J, DuBois RN (2000) Celecoxib prevents tumor growth in vivo without toxicity to normal gut: lack of correlation between in vitro and in vivo models. Cancer Res 60:6045–6051
Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S (2002) Selective cyclooxygenase-2 inhibitors show a differential ability to inhibit proliferation and induce apoptosis of colon adenocarcinoma cells. FEBS Lett 531:278–284
Arber N, DuBois RN (1999) Nonsteroidal anti-inflammatory drugs and prevention of colorectal cancer. Curr Gastroenterol Rep 1:441–448
Shiff SJ, Koutsos MI, Qiao L, Rigas B (1996) NSAIDs inhibit the proliferation of colon adeno- carcinoma cells: effects on cell cycle and apoptosis. Exp Cell Res 222:179–188
Piazza GA, Rahm AL, Krutzsch M, Sperl G, Paranka NS, Gross PH, Brendel K, Burt RW, Alberts DS, Pamukcu R, et al (1995) Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res 55:3110–3116
Dvory-Sobol H, Cohen-Noyman E, Kazanov D, Figer A, Birkenfeld S, Madar-Shapiro L, Benamouzig R, Arber N (2006) Celecoxib leads to G2/M arrest by induction of p21 and down-regulation of cyclin B1 expression in a p53-independent manner. Eur J Cancer 42:422–426
Grosch S, Tegeder I, Niederberger E, Brautigam L, Geisslinger G (2001) COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J 15:2742–2744
Orner GA, Dashwood WM, Blum CA, Diaz GD, Li Q, Dashwood RH (2003) Suppression of tumorigenesis in the Apc(min) mouse: down-regulation of beta-catenin signaling by a combination of tea plus sulindac. Carcinogenesis 24:263–267
Richter M, Weiss M, Weinberger I, Furstenberger G, Marian B (2001) Growth inhibition and induction of apoptosis in colorectal tumor cells by cyclooxygenase inhibitors. Carcinogenesis 22:17–25
Acknowledgment
The authors thank Ester Eshkol for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lev-Ari, S., Kazanov, D., Liberman, E. et al. Down-Regulation of PGE2 by Physiologic Levels of Celecoxib is not Sufficient to Induce Apoptosis or Inhibit Cell Proliferation in Human Colon Carcinoma Cell Lines. Dig Dis Sci 52, 1128–1133 (2007). https://doi.org/10.1007/s10620-006-9619-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-006-9619-x