
Abstract
The therapeutic potential of adult stem cells may become a relevant option in clinical care in the future. In hand and plastic surgery, cell therapy might be used to enhance nerve regeneration and help surgeons and clinicians to repair debilitating nerve injuries. Adipose-derived stem cells (ASCs) are found in abundant quantities and can be harvested with a low morbidity. In order to define the optimal fat harvest location and detect any potential differences in ASC proliferation properties, we compared biopsies from different anatomical sites (inguinal, flank, pericardiac, omentum, neck) in Sprague–Dawley rats. ASCs were expanded from each biopsy and a proliferation assay using different mitogenic factors, basic fibroblast growth factor (bFGF) and platelet-derived growth factor (PDGF) was performed. Our results show that when compared with the pericardiac region, cells isolated from the inguinal, flank, omental and neck regions grow significantly better in growth medium alone. bFGF significantly enhanced the growth rate of ASCs isolated from all regions except the omentum. PDGF had minimal effect on ASC proliferation rate but increases the growth of ASCs from the neck region. Analysis of all the data suggests that ASCs from the neck region may be the ideal stem cell sources for tissue engineering approaches for the regeneration of nervous tissue.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bone marrow stromal cells are known as multipotent stem cells under specific conditions (Prockop 1997) and are an attractive cell source for tissue engineering (Dezawa et al. 2001). However, the isolation of these stem cells is associated with a considerable morbidity. In general, the site of biopsy should be easily accessible, show minimal donor site morbidity and have the option of repeated biopsies if needed.
Adipose-derived stem cells (ASCs) are found in abundant quantities, have high proliferation rates, can be harvested by minimally invasive procedures and are multi-potent cells capable of becoming bone, cartilage, muscle, endovascular, neuronal, and other cells (Liang et al. 2011; Strem et al. 2005; Taha and Hedayati 2010; Witkowska-Zimny and Walenko 2011). The characteristics of ASCs are closely similar to those of other types of mesenchymal stem cells (MSCs) (Dhanasekaran et al. 2012; Zuk et al. 2002). They have been successfully tested in preclinical models, which opens a wide field of clinical applications (Wu et al. 2011; Zhang et al. 2012). Remarkable is that the frequency of ASCs in adipose tissue far exceeds the MSCs frequency in bone marrow, which is only 0.01–0.001 % (Strem et al. 2005). This makes ASCs a promising alternative source of MSCs for tissue engineering (Strioga et al. 2012).
The harvested fat sample should have a high density of expandable ASCs with minimal contamination of fascial tissues. Knowing the ideal biopsy location might allow the surgeon to take a smaller biopsy without restricting the proliferation rate of ASCs.
Recently we showed that human ASCs isolated from the superficial layer of abdominal fat tissue proliferate significantly faster than those from the deeper layer (Kalbermatten et al. 2011). In the superficial and deep layer, pluripotent stem cell markers oct4, nanog and also the stro-1 cell surface antigen were expressed (Kalbermatten et al. 2011).
Other factors such as the type of surgical procedure may also affect growth characteristics and the yield of ASCs. Oedayrajsingh-Varma et al. (2006) suggested that adipose tissue derived from both abdomen and hips harvested by resection or tumescent liposuction can be used for tissue-engineering purposes. It has also been shown that adipose tissue harvested from the breasts, because of presence of glandular cells, may lead to extensive variations in cell counts. Moreover it cannot be excluded that differentiated glandular cells affect ASC proliferation and differentiation (Oedayrajsingh-Varma et al. 2006).
In this research we hypothesized that harvest location is a key factor for the quality of ASCs for nerve regeneration. In order to define the optimal adipose tissue from which to harvest the ASCs, we compared fat tissue from different origins and evaluated the expansion potential with different mitogenic factors.
Materials and methods
Harvest and culture of ASCs
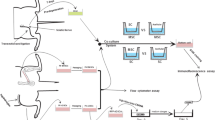
All studies were approved by the local veterinary physician. ASCs were harvested as described previously by our group (di Summa et al. 2010). One gram of adipose tissue was harvested from the most representative sources according to the literature (Aguena et al. 2012; Bayes-Genis et al. 2012; Mohammadi et al. 2012; Padoin et al. 2008; Yuan et al. 2010) (inguinal, flank, pericardiac, neck and omentum) in four male Sprague–Dawley (SD) rats aged 8 weeks weighing approximately 250 g. SD rats were selected because they are widely used in the field of tissue engineering and peripheral nerve repair (di Summa et al. 2010). The adipose tissue was carefully dissected, minced by using a sterile razor blade and placed for 2 h at 37 °C in 15 ml Hank’s balanced salt solution (HBSS) containing 0.2 % (w/v) collagenase type 1 and 20 μl penicillin/streptomycin for enzymatic digestion. Next, the enzyme was neutralized by adding growth medium (GM) containing Modified Eagle’s Medium (α-MEM; Invitrogen, Paisley, UK), 10 % (v/v) fetal bovine serum (FBS) and 1 % (v/v) penicillin/streptomycin (Gibco, Grand Island, NY, USA). The samples were centrifuged at 1,200g for 5 min and the resultant stromal cell pellet was resuspended in GM. Subsequently, the solution was passed through a 70-μm filter to remove remaining undissociated tissue, centrifuged at 1,000g for 5 min and the pellet resuspended in GM. The ASCs were propagated and plated in a 75 cm2 tissue culture flask. Cells were incubated at 37 °C with 5 % CO2 and were maintained at sub-confluent levels with passage by trypsin/EDTA (Invitrogen, UK) when required.
Cell proliferation
The proliferation of the ASCs isolated from different origins were assessed for 96 h using the CellTiter 96® Aqueous non-radioactive cell proliferation assay (Promega, Madison, WI, USA). At passage 2, 1 × 103 cells were plated in a 96-well plate using either 200 μl of GM alone, or supplemented with basic fibroblast growth factor bFGF (10 ng/ml) or platelet-derived growth factor PDGF (5 ng/ml). At the time of seeding the signal was assessed for all sources and normalized. At regular intervals (24, 48, 72, 96 h), 20 μl of Cell Titer 96® Aqueous Assay Reagents (Promega, USA) were added and incubated for 4 h at 37 °C and 5 % CO2. The absorbance was recorded at 490 nm using a spectrophotometric plate reader and measurements were calculated by subtracting the average control absorbance (medium only) from the average cell absorbance. The different patterns of proliferation among different time-points were performed in the same groups along the 96 h and compared with the 24 h time-point.
Based on the fact that the number of cells is proportional to the absorbance recorded by the Cell Titer Assay we were able to calculate population doubling (PD) times using exponential curve fitting software (Mohamet et al. 2010).
Immunocytochemistry
ASCs (2 × 104 cells) at passage 2 were plated on Lab-Tek 4-well chamber slides (Thermo Scientific, Nunc, Fisher Scientific AG, Wohlen, Switzerland), cultured for 3 days until confluence of 60 % was reached, washed with phosphate buffered saline (PBS) and fixed for 20 min in 4 % paraformaldehyde. The cells were washed for 2 min in PBS and blocked for 1 h in immunofluorescence blocking buffer (IBB) containing 1 % bovine serum albumin (BSA, Invitrogen, 15561-020), 1× PBS and 0.1 % Triton X-100 (Sigma-Aldrich, Munich, Germany). After washing three times with PBS the cells were subsequently incubated over night at 4° in PBS with 0.1 % (v/v) Triton containing the following antibodies: S100 (Z0311, Dako, Baar, Switzerland) (1:500), Nestin (MAB353, Chemicon, Temecula, CA, USA) (1:200) and GFAP (Neomarkers Ab-1, Clone GA-5, Lab Vision/Thermo Fisher Scientific) (1:400). The cells were washed three times with PBS and incubated for 40 min under light protection in PBS containing Alexa Fluor® 488 goat anti-rabbit IgG (Invitrogen, A-11008) (1:500), Alexa Fluor® 594 goat anti-mouse IgG (Invitrogen, A-11005) (1:500) or Alexa Fluor® 488 goat anti-mouse IgG (Invitrogen, A-11001) (1:500). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (Sigma, D9542) (1:500).
Statistical analysis
Data are presented as mean ± SEM from four animals. All experimental samples were conducted in triplicate and the four rats were used as independent biological replicates. Where appropriate t-tests or one-way ANOVA with Bonferoni post hoc test for multiple comparisons were used to determine the statistically significant differences between groups. The following convention was used in the figures: *p < 0.05; **p < 0.01; ***p < 0.0001.
Results
One gram of adipose tissue biopsy was harvested from different origins (inguinal, flank, pericardiac, neck and omentum) (Fig. 1) in rats. After enzymatic digestion and centrifugation the stromal cell fraction was isolated from mature adipocytes and plated in a 75-cm2 flask. After 3 weeks in culture, upon reaching confluence, cells were trypsinized and counted. ASCs were plastic-adherent and expressed basal levels of S100 with a high expression in ASCs from the neck and flank regions (Fig. 2b–f). A small fraction of cells was found to be nestin positive (Fig. 2a). Undifferentiated ASCs were negative for expression of GFAP (data not shown). We compared the proliferation of the ASCs (passage 2) isolated from over a 96 h period. Within 96 h the cells were still proliferating and importantly did not show a saturating signal in the assay. The cells were incubated with either GM alone or GM with bFGF or PDGF.

Different harvest locations with harvest tissue aside A neck, B flank, C inguinal, D pericardiac, E omentum

Representative immunofluorescent staining of ASCs from different harvest locations. The cells were stained with nestin (red) and S100 (green), respectively. The nuclei are stained with DAPI (4′-6-diamidino-2-phenylindole, blue). Scale bar 100 μm. (Color figure online)
As shown in Fig. 3a, ASCs isolated from the inguinal, flank and omental region grow significantly better after 96 h in GM alone versus after 24 h. ASCs from the neck region grow least in GM. However, bFGF significantly enhanced the growth rate of all types within 48 h, with the greatest effect on neck ASCs. On the other hand, ASCs from the pericardiac and omental regions responded least (Fig. 3b). Similar results were observed with PDGF; PDGF speeded the proliferation of ASCs from the neck region significantly better within 72 h but had no effect on ASCs from the pericardiac region (Fig. 3c). From proliferation assays, PD times were calculated for the cells at passage 2. Cells isolated from the inguinal, flank, omental and neck regions all showed similar PD times whereas ASCs extracted from pericardiac tissue had a significantly longer PD time (Fig. 4). Treatment with bFGF reduced PD time for all the cultures except the cells isolated from the omentum (Fig. 4). In contrast, application of PDGF only reduced the PD time of ASCs taken from the neck region (Fig. 4).

Proliferation rate of ASC from different origins (ing inguinal, flk flank, peri pericardiac, om omental, and neck) with GM alone (a), or addition of bFGF (b) and PDGF (c). *p < 0.05, **p < 0.01, ***p < 0.001 significantly different from respective samples at 24 h and # p < 0.05 significantly different at 96 h compared with ASCs from inguinal, flank or neck regions

Population doubling (PD) times for ASCs isolated from the different harvest locations and cultured in growth medium alone (GM) or in the presence of bFGF or PDGF. *p < 0.05, **p < 0.01 significantly different from pericardiac ASCs. # p < 0.05 significantly decreased PD time in the presence of bFGF
Discussion
Nerve injury is very common and often affecting young patients (mean age 30). In spite of advanced treatment options involving microsurgery, almost all patients are left with lifelong disabilities involving loss of sensation and motor function (Lundborg 2000). New tissue-engineering approaches might facilitate the process of nerve repair and regeneration and would certainly represent a tremendous medical breakthrough. An advantageous approach for clinical practice would be to harvest ASCs and immediately give them back to the patient within the same operation, the so called “one-step surgical procedure” (Helder et al. 2007).
As ASCs from different origins might have different utility for the treatment of nerve injury, we investigated whether cell proliferation differs among different origins. For this purpose we used a model system which keeps other environmental factors stable, including age and donor gender.
Our results showed that pericardiac ASCs grew significantly slower than the cells isolated from other regions. The growth rate was enhanced for all types except omental ASCs with bFGF. PDGF had minimal effect on ASCs proliferation rate, only enhancing the growth of ASCs from the neck region. This goes in line with our immunohistochemical analysis where high levels of S100 expression were found in ASCs from the flank and neck regions. Furthermore, a basal expression of nestin but no expression of GFAP was found. This is consistent with the literature where undifferentiated ASCs have been found to constitutively express neural specific proteins (Deng et al. 2006; Kaewkhaw et al. 2011; Lattanzi et al. 2011). It is suggested that nestin expression is an essential prerequisite for MSCs to progress toward a neural lineage (Wislet-Gendebien et al. 2003).
Controversy exists about the capacity of MSCs to transdifferentiate into lineages derived from the neuroectoderm (Phinney and Prockop 2007). New findings suggest that the ability of MSCs to alter the tissue microenvironment via secretion of soluble factors may contribute more significantly than their capacity for transdifferentiation in tissue repair (Phinney and Prockop 2007). In addition, stromal cells have been shown to express neurotrophic receptor proteins (Labouyrie et al. 1999). Thus, our results could be attributed to the fact that the various populations of ASCs could express different levels of basic FGF receptor or contain CD271-expressing cells, which have been shown to co-express CD140b (platelet-derived growth factor receptor β (Buhring et al. 2007).
In this study we decided to use bFGF and PDGF as they are potent mitogens for glial cells and have a potential effect on nerve repair (Lutton et al. 2012; Yun et al. 2010). Fibroblast growth factors (FGFs) have shown potential effects on the repair and regeneration of a wide spectrum of tissues, including skin, blood vessel, muscle (Tassi et al. 2011), de novo adipogenesis (Tabata et al. 2000), tendon/ligament (Hankemeier et al. 2005), cartilage (Martin et al. 2001), bone (Kimura et al. 2008), tooth (Kitamura et al. 2011), and nerve tissues (Yun et al. 2010).
PDGF was first identified as a factor in platelets which allowed the growth of fibroblasts in vitro (Kohler and Lipton 1974). Further characterization of this factor demonstrated that it is a potent mitogen for all cells of mesenchymal origin, including vascular smooth muscle cells and glial cells (Ross et al. 1986). PDGF is, as FGF, an angiogenic growth factor that accelerates capillary formation (Sato et al. 1993). Adipose tissue growth seems to be regulated through angiogenesis (Hausman and Richardson 2004) and PDGF itself stimulates pre-adipocyte proliferation (Hauner et al. 1995). Furthermore, it has been reported that a mixture of glial growth factors (containing heregulin, forskolin, PDGF and bFGF) can induce ASCs into cells which are spindle-like in shape and express GFAP, S100 and p75, similar to genuine Schwann cells (SCs) (Jiang et al. 2008; Kingham et al. 2007). In response to forskolin, cultured primary SCs increase cAMP and hence myelin protein p0 can be induced (Morgan et al. 1991). Expression of p0 in response to forskolin is a very important feature of SCs, which indicates that SCs may have myelin-forming ability (Morgan et al. 1991). In addition, cAMP elevation is reported to enhance the responsiveness of cells to trophic factors (Meyer-Franke et al. 1998). Different mitogenic factors, amongst others bFGF and PDGF could have a synergistic effect in enhancement of these factors to MSCs (Dezawa et al. 2001).
Finally, our study has some limitations since the cells we investigated were isolated from experimental animals. Thus, due to potential differences between human and rodent MSCs direct comparisons cannot be drawn. Nevertheless, our results have identified that ASCs isolated from the neck region proliferate rapidly in response to bFGF and show enhanced growth with PDGF treatment. Future studies will utilise these cells in experimental in vivo models to assess their functional effects within the injured nervous system.
References
Aguena M, Fanganiello RD, Tissiani LA, Ishiy FA, Atique R, Alonso N, Passos-Bueno MR (2012) Optimization of parameters for a more efficient use of adipose-derived stem cells in regenerative medicine therapies. Stem Cells Int 2012:303610
Bayes-Genis A, Galvez-Monton C, Prat-Vidal C, Soler-Botija C (2012) Cardiac adipose tissue: a new frontier for cardiac regeneration? Int J Cardiol. doi:10.1016/j.ijcard.2012.05.082
Buhring HJ, Battula VL, Treml S, Schewe B, Kanz L, Vogel W (2007) Novel markers for the prospective isolation of human MSC. Ann N Y Acad Sci 1106:262–271
Deng J, Petersen BE, Steindler DA, Jorgensen ML, Laywell ED (2006) Mesenchymal stem cells spontaneously express neural proteins in culture and are neurogenic after transplantation. Stem Cells 24:1054–1064
Dezawa M, Takahashi I, Esaki M, Takano M, Sawada H (2001) Sciatic nerve regeneration in rats induced by transplantation of in vitro differentiated bone-marrow stromal cells. Eur J Neurosci 14:1771–1776
Dhanasekaran M, Indumathi S, Kanmani A, Poojitha R, Revathy KM, Rajkumar JS, Sudarsanam D (2012) Surface antigenic profiling of stem cells from human omentum fat in comparison with subcutaneous fat and bone marrow. Cytotechnology 5:497–509
di Summa PG, Kingham PJ, Raffoul W, Wiberg M, Terenghi G, Kalbermatten DF (2010) Adipose-derived stem cells enhance peripheral nerve regeneration. J Plast Reconstr Aesthet Surg 63:1544–1552
Hankemeier S, Keus M, Zeichen J, Jagodzinski M, Barkhausen T, Bosch U, Krettek C, Van Griensven M (2005) Modulation of proliferation and differentiation of human bone marrow stromal cells by fibroblast growth factor 2: potential implications for tissue engineering of tendons and ligaments. Tissue Eng 11:41–49
Hauner H, Rohrig K, Petruschke T (1995) Effects of epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) on human adipocyte development and function. Eur J Clin Investig 25:90–96
Hausman GJ, Richardson RL (2004) Adipose tissue angiogenesis. J Animal Sci 82:925–934
Helder MN, Knippenberg M, Klein-Nulend J, Wuisman PI (2007) Stem cells from adipose tissue allow challenging new concepts for regenerative medicine. Tissue Eng 13:1799–1808
Jiang L, Zhu JK, Liu XL, Xiang P, Hu J, Yu WH (2008) Differentiation of rat adipose tissue-derived stem cells into Schwann-like cells in vitro. NeuroReport 19:1015–1019
Kaewkhaw R, Scutt AM, Haycock JW (2011) Anatomical site influences the differentiation of adipose-derived stem cells for Schwann-cell phenotype and function. Glia 59:734–749
Kalbermatten DF, Schaakxs D, Kingham PJ, Wiberg M (2011) Neurotrophic activity of human adipose stem cells isolated from deep and superficial layers of abdominal fat. Cell Tissue Res 344:251–260
Kimura Y, Hokugo A, Takamoto T, Tabata Y, Kurosawa H (2008) Regeneration of anterior cruciate ligament by biodegradable scaffold combined with local controlled release of basic fibroblast growth factor and collagen wrapping. Tissue Eng Part C Methods 14:47–57
Kingham PJ, Kalbermatten DF, Mahay D, Armstrong SJ, Wiberg M, Terenghi G (2007) Adipose-derived stem cells differentiate into a Schwann cell phenotype and promote neurite outgrowth in vitro. Exp Neurol 207:267–274
Kitamura M, Akamatsu M, Machigashira M, Hara Y, Sakagami R, Hirofuji T, Hamachi T, Maeda K, Yokota M, Kido J, Nagata T, Kurihara H, Takashiba S, Sibutani T, Fukuda M, Noguchi T, Yamazaki K, Yoshie H, Ioroi K, Arai T, Nakagawa T, Ito K, Oda S, Izumi Y, Ogata Y, Yamada S, Shimauchi H, Kunimatsu K, Kawanami M, Fujii T, Furuichi Y, Furuuchi T, Sasano T, Imai E, Omae M, Yamada S, Watanuki M, Murakami S (2011) FGF-2 stimulates periodontal regeneration: results of a multi-center randomized clinical trial. J Dent Res 90:35–40
Kohler N, Lipton A (1974) Platelets as a source of fibroblast growth-promoting activity. Exp Cell Res 87:297–301
Labouyrie E, Dubus P, Groppi A, Mahon FX, Ferrer J, Parrens M, Reiffers J, de Mascarel A, Merlio JP (1999) Expression of neurotrophins and their receptors in human bone marrow. Am J Pathol 154:405–415
Lattanzi W, Geloso MC, Saulnier N, Giannetti S, Puglisi MA, Corvino V, Gasbarrini A, Michetti F (2011) Neurotrophic features of human adipose tissue-derived stromal cells: in vitro and in vivo studies. J Biomed Biotechnol 2011:468705
Liang W, Xia H, Li J, Zhao RC (2011) Human adipose tissue derived mesenchymal stem cells are resistant to several chemotherapeutic agents. Cytotechnology 63:523–530
Lundborg G (2000) A 25-year perspective of peripheral nerve surgery: evolving neuroscientific concepts and clinical significance. J Hand Surg Am 25:391–414
Lutton C, Young YW, Williams R, Meedeniya AC, Mackay-Sim A, Goss B (2012) Combined VEGF and PDGF treatment reduces secondary degeneration after spinal cord injury. J Neurotrauma 29:957–970
Martin I, Suetterlin R, Baschong W, Heberer M, Vunjak-Novakovic G, Freed LE (2001) Enhanced cartilage tissue engineering by sequential exposure of chondrocytes to FGF-2 during 2D expansion and BMP-2 during 3D cultivation. J Cell Biochem 83:121–128
Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG Jr, Reichardt LF, Barres BA (1998) Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21:681–693
Mohamet L, Lea ML, Ward CM (2010) Abrogation of E-cadherin-mediated cellular aggregation allows proliferation of pluripotent mouse embryonic stem cells in shake flask bioreactors. PLoS ONE 5:e12921
Mohammadi R, Azizi S, Amini K (2012) Effects of undifferentiated cultured omental adipose-derived stem cells on peripheral nerve regeneration. J Surg Res. doi:10.1016/j.jss.2012.04.011
Morgan L, Jessen KR, Mirsky R (1991) The effects of cAMP on differentiation of cultured Schwann cells: progression from an early phenotype (04+) to a myelin phenotype (P0+, GFAP-, N-CAM-, NGF-receptor-) depends on growth inhibition. J Cell Biol 112:457–467
Oedayrajsingh-Varma MJ, van Ham SM, Knippenberg M, Helder MN, Klein-Nulend J, Schouten TE, Ritt MJ, van Milligen FJ (2006) Adipose tissue-derived mesenchymal stem cell yield and growth characteristics are affected by the tissue-harvesting procedure. Cytotherapy 8:166–177
Padoin AV, Braga-Silva J, Martins P, Rezende K, Rezende AR, Grechi B, Gehlen D, Machado DC (2008) Sources of processed lipoaspirate cells: influence of donor site on cell concentration. Plast Reconstr Surg 122:614–618
Phinney DG, Prockop DJ (2007) Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair–current views. Stem Cells 25:2896–2902
Prockop DJ (1997) Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 276:71–74
Ross R, Raines EW, Bowen-Pope DF (1986) The biology of platelet-derived growth factor. Cell 46:155–169
Sato N, Beitz JG, Kato J, Yamamoto M, Clark JW, Calabresi P, Raymond A, Frackelton AR Jr (1993) Platelet-derived growth factor indirectly stimulates angiogenesis in vitro. Am J Pathol 142:1119–1130
Strem BM, Hicok KC, Zhu M, Wulur I, Alfonso Z, Schreiber RE, Fraser JK, Hedrick MH (2005) Multipotential differentiation of adipose tissue-derived stem cells. Keio J Med 54:132–141
Strioga M, Viswanathan S, Darinskas A, Slaby O, Michalek J (2012) Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev 21:2724–2752
Tabata Y, Miyao M, Inamoto T, Ishii T, Hirano Y, Yamaoki Y, Ikada Y (2000) De novo formation of adipose tissue by controlled release of basic fibroblast growth factor. Tissue Eng 6:279–289
Taha MF, Hedayati V (2010) Isolation, identification and multipotential differentiation of mouse adipose tissue-derived stem cells. Tissue Cell 42:211–216
Tassi E, McDonnell K, Gibby KA, Tilan JU, Kim SE, Kodack DP, Schmidt MO, Sharif GM, Wilcox CS, Welch WJ, Gallicano GI, Johnson MD, Riegel AT, Wellstein A (2011) Impact of fibroblast growth factor-binding protein-1 expression on angiogenesis and wound healing. Am J Pathol 179:2220–2232
Wislet-Gendebien S, Leprince P, Moonen G, Rogister B (2003) Regulation of neural markers nestin and GFAP expression by cultivated bone marrow stromal cells. J Cell Sci 116:3295–3302
Witkowska-Zimny M, Walenko K (2011) Stem cells from adipose tissue. Cell Mol Biol Lett 16:236–257
Wu G, Song Y, Zheng X, Jiang Z (2011) Adipose-derived stromal cell transplantation for treatment of stress urinary incontinence. Tissue Cell 43:246–253
Yuan Q, Zeng X, Chen L, Peng E, Ye Z (2010) Comparison of myogenic differentiation ability of adipose-derived stem cells from different sites in rabbit. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 24:1228–1232
Yun YR, Won JE, Jeon E, Lee S, Kang W, Jo H, Jang JH, Shin US, Kim HW (2010) Fibroblast growth factors: biology, function, and application for tissue regeneration. J Tissue Eng 2010:218142
Zhang Y, Wang F, Chen J, Ning Z, Yang L (2012) Bone marrow-derived mesenchymal stem cells versus bone marrow nucleated cells in the treatment of chondral defects. Int Orthop 36:1079–1086
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH (2002) Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13:4279–4295
Conflict of interest
No competing financial interests exist.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Patricia E. Engels and Mathias Tremp contributed equally to this work.
Rights and permissions
About this article
Cite this article
Engels, P.E., Tremp, M., Kingham, P.J. et al. Harvest site influences the growth properties of adipose derived stem cells. Cytotechnology 65, 437–445 (2013). https://doi.org/10.1007/s10616-012-9498-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-012-9498-2