
Sequential transformations of dipterocarpol (oximation, first-order Beckmann rearrangement, dehydration, isomerization) synthesized the new dammarane-type triterpenoid A-azepanodammara-20(21),24(25)-diene. Its cytotoxicity was studied in vitro against 60 cell lines of 9 human tumor types. The cytotoxicity against all cancer cell types increased significantly as compared to native dipterocarpol if an azepane fragment was introduced into ring A (percent inhibition from 15.72% to 82.08%). Detailed cytotoxicity studies confirmed the activity against a broad spectrum of human tumor cell lines with a mean (MID) log LC50 of 5.57.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Triterpenoids from plants are an important source of starting materials for synthesizing various structural derivatives and studying their anticancer activity. Thus, diosphenols of 23-hydroxybetulonic acid are candidates for in vivo tests on melanoma and liver cancer in mouse models. Hydrazides and hydrazones of betulonic acid were highly cytotoxic to endothelial cell lines ECV304 [1, 2]. The leading dammaranes are derivatives of protopanaxadiol, the aglycon of ginseng root glycosides with broad spectra of cytotoxicity. For example, 25-methoxydammara-3β,12β,20-triol and (20S)-25-ethoxy-3,12,20-trihydroxydammar-23-ene were active against prostate, liver, and breast cancer; leukemia; and neuroblastoma [3, 4].
Azepane rings are important biologically active heterocycles [5]. They were present in triterpenoids with anticancer, antituberculosis, and antidiabetic activity [6,7,8]. Herein, the first synthesis of a dammarane triterpenoid with an A-azepane ring is reported. The starting compound was dipterocarpol, the main metabolite in sap of the tropical tree Dipterocarpus alatus, which possessed anticancer [9], antiviral [10,11,12,13], immunostimulatory [14], and other types of activity.

Survival of cells cultivated with test compounds (100 μM) in percent vs. control cells (without test compounds added to growth medium) are given. Negative values correspond to cell death. A “–“ sign means no data.
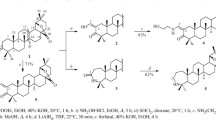
According to the literature [15], the reaction of dipterocarpol (1) with NH2OH·HCl in Py produced oxime 2, refluxing of which in AcOH dehydrated the hydroxy and formed 3 with a 20(21)-double bond in the side chain. An analogous process was observed by us during preparation of hollongdione [16]. According to PMR spectra, 3-hydroxyiminodammara-20(21),24(25)-diene (2) was formed only as the syn-stereoisomer. The resonance for H-21 was observed as a doublet at δ 4.72 ppm in the spectrum of 3. The 13C NMR spectrum showed a characteristic resonance for C3 at δ 166.8 ppm; for the C-20(21) double bond, 152.5 and 107.6. A first-order Beckmann rearrangement of 3 by treatment with SOCl2 in dioxane gave azepane 4 in 65% yield. These transformations caused changes in the NMR spectra. The resonance for C-3 was observed at δ 176.3 ppm; for the NH proton, as a broad singlet at δ 5.51 ppm. Compound 4 was reduced by LiAlH4 in refluxing THF to give 5 with an azepane ring A in 62% yield after chromatographic purification.
Antitumor activity in vitro (cytotoxicity) of 1 and derivatives 3–5 was studied using 60 cell lines of 9 different human tumors (lung, colon, CNS, ovary, kidney, prostate, brain, leukemia, melanoma) according to the National Cancer Institute (NCI, USA) method [17,18,19,20]. Table 1 presents the percent growth of the treated cells as compared to control cells (negative values correspond to cell death). Compounds were considered active for cell growth inhibition up to 32% of the control or their death according to criteria adopted by the NCI. Table 1 shows that starting 1 and 3-syn-hydroxyiminodammara-20(21),24(25)-diene (3) were inactive. Azepanone 4 and azepane 5 displayed broad spectra of activity against several tumor cells. For example, cells of eight human tumors were sensitive to 4. The greatest activity was observed against two cancer cell types, i.e., melanoma (LOX IMVI, MALME-3M, SK-MEL-5) and colon (UO-31, SN12C, RXF393). Azepane 5 exhibited antitumor activity against nine cancer cell types with an inhibition range from 15.72% to 82.08%. Compound 5 was cytocidal for 58 of the 60 tested tumor cell lines.
Detailed studies of 4 and 5 confirmed their significant cytotoxicities against broad spectra of human tumor cell lines. In general, the cytotoxicity of 5 was greater than that of 4. The log LC50 for 5 was 5.57 (MID); for 4, 4.71 (MID).
Thus, the synthesis of the new triterpenoid A-azepanodammara-20(21),24(25)-diene with high cytotoxicity is reported.
Experimental
PMR and 13C NMR spectra were recorded in CDCl3 with TMS internal standard on a Bruker AM-300 spectrometer (Germany) at 300 and 75.5 MHz using equipment at the Khimiya CUC. Melting points were determined on a Boetius apparatus. Optical rotation was measured in a 1-dm tube on a PerkinElmer 241 MC polarimeter. TLC analysis used Sorbfil plates (ZAO Sorbpolimer, Russia) and CHCl3–EtOAc (40:1) with detection by H2SO4 solution (10%, 2–3 min at 100–120°C). Elemental analyses were performed on a Euro EA 3000 CHNS analyzer using acetanilide as a primary standard. Column chromatography used neutral Al2O3 (Reakhim). Dipterocarpol (1) was isolated from D. alatus sap as before [21]. Spectral characteristics of 2 were published [15].
Logarithms of molar concentrations causing 50% cell growth inhibition (log GI50), total cell growth inhibition (log TGI), and 50% cell death (LC50) are given. If these compound concentrations exceeded 100 μM, then their exact values were not calculated and are shown in the table as “> –4.00”. A “–“ sign means no data.
3-syn-Hydroxyiminodammara-20(12),24(25)-diene (3). Compound 2 (0.46 g, 1 mmol) was dissolved in AcOH (20 mL), stirred at 40°C for 2 h, and poured into H2O (20 mL). The precipitate was filtered off, rinsed with H2O to pH 7, dried in air, and purified by column chromatography using n-C6H12–EtOAc (4:1). Yield 0.38 g (87%), mp 132–133°C, Rf 0.75, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +130° (c 0.1, CHCl3). C30H49NO. 1H NMR spectrum (300 MHz, CDCl3, δ, ppm, J/Hz): 0.83, 0.95, 1.02, 1.08, 1.15 (3H each, s, CH3), 1.21–1.62 (14H, m, CH, CH2), 1.64 (3H, s, CH3), 1.69 (3H, s, CH3), 1.83–2.01 (8H, m, CH, CH2), 2.02–2.79 (3H, m, CH, CH2), 4.72 (1H, d, J = 8.6, H-21), 5.13 (1H, t, J = 7.1, H-24), 7.71 (1H, br.s, NH). 13C NMR spectrum (75 MHz, CDCl3, δ, ppm): 15.3, 15.8, 16.3, 17.2, 17.6, 19.0, 21.7, 22.5, 22.8, 24.7, 25.4, 25.7, 27.3, 27.5, 31.1, 34.8, 37.1, 39.0, 40.4, 40.5, 42.3, 42.6, 49.7, 50.2, 55.4, 107.6 (C-21), 124.5 (C-24), 131.5 (C-25), 152.5 (C-20), 166.8 (C-3).
3-Oxo-3a-homo-3a-azadammara-20(21),24(25)-diene (4). A solution of 3 (0.44 g, 1 mmol) in anhydrous dioxane (15 mL) was treated with SOCl2 (0.4 mL), stirred at room temperature for 30 min, and poured into H2O (50 mL). The precipitate was filtered off, rinsed with H2O to pH 7, dried in air, and purified by column chromatography using n-C6H12, n-C6H12–EtOAc (4:1), and CHCl3. Yield 0.32 g (75%), mp 97°C, Rf 0.30, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +122° (c 0.1, CHCl3). C30H49NO. 1H NMR spectrum (300 MHz, CDCl3, δ, ppm, J/Hz): 0.84, 1.01, 1.06, 1.22, 1.32 (3H each, s, CH3), 1.33–1.56 (14H, m, CH, CH2), 1.62 (3H, s, CH3), 1.68 (3H, s, CH3), 1.80–2.65 (8H, m, CH, CH2), 4.72 (1H, d, J = 13.8, H-21), 5.12 (1H, t, J = 7.0, H-24), 5.51 (1H, br.s, NH). 13C NMR spectrum (75 MHz, CDCl3, δ, ppm): 15.3, 15.6, 17.7, 18.3, 22.5, 22.6, 25.2, 25.7, 27.0, 27.5, 28.8, 31.2, 32.1, 33.3, 34.1, 34.8, 39.7, 40.2, 40.3, 45.5, 47.7, 49.4, 51.5, 53.3, 56.3, 107.6 (C-21), 124.4 (C-24), 131.5 (C-25), 152.5 (C-20), 176.3 (C-3).
3a-Homo-3a-azadammara-20(21),24(25)-diene (5). A solution of 4 (0.44 g, 1 mmol) in anhydrous THF (20 mL) was treated with LiAlH4 (0.046 g, 1.3 mmol), refluxed for 3 h, poured into HCl solution (5%, 200 mL), and extracted with CHCl3 (2 . 20 mL). The organic layer was separated, washed with H2O (2 . 50 mL), dried over CaCl2, evaporated in vacuo, and purified by column chromatography using CHCl3 and CHCl3–EtOH (100:1). Yield 0.27 g (62%), mp 102°C, Rf 0.23, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +114° (c 0.1, CHCl3). C30H51N. 1H NMR spectrum (300 MHz, CDCl3, δ, ppm, J/Hz): 0.89, 0.91, 1.06, 1.19, 1.22, 1.45 (3H each, s, CH3), 1.19–1.45 (14H, m, CH, CH2), 1.63 (3H, s, CH3), 1.71 (3H, s, CH3), 1.75–3.15 (14H, m, CH, CH2), 4.72 (1H, d, J = 8.6, H-21), 5.12 (1H, t, J = 7.0, H-24). 13C NMR spectrum (75 MHz, CDCl3, δ, ppm): 15.5, 15.9, 16.3, 17.7, 21.4, 22.0, 23.2, 25.2, 27.0, 27.3, 27.9, 28.9, 31.5, 34.1, 34.8, 39.9, 40.3, 40.7, 41.3, 45.6, 47.7, 49.5, 49.7, 53.3, 55.1, 63.2 (C-3), 107.7 (C-21), 124.4 (C-24), 131.4 (C-25), 152.4 (C-20).
The test method forin vitroantitumor activity of 1 and 3–5 against 60 cell lines of 9 different human tumors is available at https://dtp.cancer.gov/discovery_develoment/nci-60/methodology.htm. All compounds were dissolved in DMSO for the screening. Detailed methods for preparing aliquots of compounds are given at the site https://dtp.cancer.gov/discovery_development/nci-60/handling.htm. Preliminary testing used a single compound concentration of 10–5 M in cell growth medium for 48 h, after which growth of treated cells was estimated by comparisons with untreated control cells.
Compounds showing significant growth inhibition in the single-dose screening were evaluated by comparing them using 60-cell panels and five concentrations.
Compounds 4 and 5 showed significant cytotoxicity in preliminary screening and were tested at five different concentrations (Table 2). The logarithms of the molar concentrations causing 50% growth inhibition of the cells (log GI50), total growth inhibition (log TGI), and death of 50% of the cells (log LC50) were calculated from the results. Lower values of log GI50, log TGI, and log LC50 were indicative of cytotoxic effects at lower concentrations, i.e., of higher cytotoxicity.
Cells were cultivated in RPMI-1640 medium with added bovine serum (5%) and L-glutamine (2 mM) in 96-well plates for 24 h. Then, test compound for preliminary testing was added to a concentration of 100 μM. Cells were rinsed after 48 h to remove the test compounds and were treated with sulforhodamine B dye for 10 min. Dye bound by living cells was dissolved. The optical density of the resulting solution was proportional to the number of living cells. The optical density was measured by photometry (using seven measurements) at 515 nm for the dye in cell cultures after cultivation for 48 h in the presence of the compounds (Ti), in control cell cultures from time z before adding the compounds to the test cultures (Tz), and in control cultures from time z for 48 h without test compounds (C). Three dose–response parameters were calculated for each compound using the formulas
50% percent growth inhibition (GI50):
The drug concentration causing total growth inhibition (TGI):
LC50: [(Ti – Tz)/Tz] 100 for cases where Ti < Tz. (cases where compounds induce cell death).
The reference drug was Adriamycin (doxorubicin), NCI 123127.
Detailed information about the cell line collections of the NCI is available upon request at https://dtp.cancer.gov/discovery_development/nci-60/cell_list.htm.
References
H. Zhang, P. Zhu, J. Liu, X. Yang, S. Xu, H. Yao, J. Jiang, W. Ye, X. Wu, and J. Xu, Eur. J. Med. Chem., 87, 159 (2014).
R. Mukherjee, M. Jaggi, P. Rajendran, M. J. Siddiqui, S. K. Srivastava, A. Vardhan, and A. C. Burman, Bioorg. Med. Chem. Lett., 14, 2181 (2004).
R. W. Zhang, W. Wang, H. Wang, and Y. Zhao, “Novel Ginsenoside Compounds, Compositions, and Methods of Use,” WO076900A1 (2008).
G. Chen, W. Song, X. Yang, H. Ge, and J. Li, “Protopanaxadiol Derivative and Application Thereof,” CN103387596A (2013).
G.-F. Zha, K. P. Rakesh, H. M. Manukumar, C. S. Shantharam, and S. E. Long, J. Med. Chem., 162, 465 (2019).
N. I. Medvedeva, O. B. Kazakova, T. V. Lopatina, I. E. Smirnova, G. V. Giniyatullina, I. P. Baikova, and V. E. Kataev, J. Med. Chem., 143, 464 (2018).
O. B. Kazakova, G. V. Giniyatullina, N. I. Medvedeva, T. V. Lopatina, I. P. Baikova, G. A. Tolstikov, and G. N. Apryshko, Bioorg. Khim., 40, 217 (2014).
E. F. Khusnutdinova, I. E. Smirnova, G. V. Giniyatullina, N. I. Medvedeva, E. Y. Yamansarov, D. V. Kazakov, O. B. Kazakova, P. T. Linh, Q. Viet, and D. T. Huong, Nat. Prod. Commun., 11, 33 (2016).
M. Ukiya, T. Kikuchi, H. Tokuda, K. Tabata, Y. Kimura, T. Arai, Y. Ezaki, O. Oseto, T. Suzuki, and T. Akihisha, Chem. Biodiversity, 7, 1871 (2017).
T. Akihisia, H. Tokuda, M. Ukiya, T. Suzuki, Enjo, K. Koike, T. Nikado, and H. Nishino, Chem. Pharm. Bull., 52, 153 (2004).
K. Hjrabayashi, S. Iwata, and H. Matsumoto, Chem. Pharm. Bull., 39, 112 (1991).
F. Inada, M. Somekawa, and H. Murata, Chem. Pharm. Bull., 41, 617 (1993).
G. V. Platonov, A. D. Zorina, M. A. Gordon, N. P. Chizhov, L. V. Balykina, Yu. D. Mikhailov, D. R. Ivanen, Tran Kim Kvi, and A. G. Shavva, Khim.-farm. Zh., 2, 42 (1995).
L. Revesz, P. Hiestand, L. La Vecchia, R. Naef, H.-U. Naegeli, L. Oberer, and H.-J. Roth, Bioorg. Med. Chem. Lett., 9, 1521 (1999).
Do Thi Thu Huong, Tran Thi Thu Thuy, Tran Thi Hien, Nguyen Thanh Tra, Nguyen Quyet Tien, I. E. Smirnova, O. B. Kazakova, E. M. Minnibaeva, and G. A. Tolstikov, Chem. Nat. Compd., 49, 58 (2013).
I. E. Smirnova, O. B. Kazakova, D. T. T. Huong, E. M. Minnibaeva, A. N. Lobov, and K. Yu. Suponitsky, Nat. Prod. Commun., 9, 1417 (2014).
M. C. Alley, D. A. Scudiero, P. A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker, and M. R. Boyd, Cancer Res., 48 (3), 589 (1988).
M. R. Grever, S. A. Schepartz, and B. A. Chabner, Semin. Oncol., 19, 622 (1992).
M. R. Boyd and K. D. Paull, Drug Dev., 34, 91 (1995).
R. H. Shoemaker, Nat. Rev. Cancer, 6, 813 (2006).
L. Q. Tran and K. Q. Tran, J. Chem., 36, 8 (1998).
Acknowledgment
The work was performed on State Task Topic No. AAAA-A17-117011910023-2. We thank the National Cancer Institute for determining the in vitro antitumor activity of 1 and 3–5.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2019, pp. 760–765.
Rights and permissions
About this article

Cite this article
Smirnova, I.E., Petrova, A.V. & Kazakova, O.B. Synthesis and Cytotoxicity of A-Azepanodammaradiene. Chem Nat Compd 55, 883–889 (2019). https://doi.org/10.1007/s10600-019-02838-w
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-019-02838-w