
New derivatives with hydroxy, epoxy, hydroxyimino, acetoxy, lactol, methoxylactol, and indole groups in ring A and the side chain were synthesized via chemical transformations of dipterocarpol. The structure– cytotoxicity relationship was described for the dipterocarpol derivatives.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Dipterocarpol [1, 20(S)-dammar-24-en-3-one)], a dammarane-type triterpenoid, and its derivatives exhibit various types of activity (anticancer [1], antiviral [2–4], immunostimulating [5], etc.). Recent studies have found antitumor activity of dammarane metabolites from Panax ginseng root with IC50 from 4 to 8 fM against HepG2, Colon205, and HL-60 cells [6, 7]. Metabolites of fruit and leaves of Aglaia erythrosperma exhibited cytotoxicity with IC50 from 5 to 8 μM against epidermoid carcinoma KB, breast cancer BC, and lung cancer NCI-H187 cells [8]. Conjugates of dammaranic acid with various L-amino acids were active against melanoma CRL1579 (EC50 7.5–14.5 μM) and leukemia HL60 (EC50 4.7–21.7 μM) cells [9]. Synthetically produced 20(S)-20-hydroxy-3,4-seco-dammara-4(28),24-dien-3-al inhibited carcinogenesis (skin cancer) in mice [9]. The cytotoxicity of dammarane triterpenoids depends to a large extent on the number and location of hydroxyls in their structures. For example, 7β-hydroxydipterocarpol exhibited cytotoxicity against HeLa and COS-1 cells (IC50 100 and 200 μM) whereas 7/,11α-dihydroxydipterocarpol was inactive [10]. The aglycons of ginseng root saponins 20(S)-protopanaxadiol and 20(S)-protopanaxatriol are close structural analogs of 1 that were approved as the preparation Pandimex in China for treating metastatic cancer of the lung, breast, spleen, stomach, and rectum. These dammarane saponins cause apoptosis of cells and inhibit P-glycoprotein [11]. Thus, chemical modification of 1 and the study of the cytotoxicity of its derivatives are highly critical.
Herein we present results on a series of new transformations of 1 that was isolated from sap of the tree Dipterocarpus alatus growing in Vietnam.
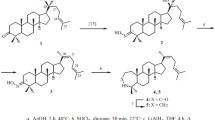
Oxidation of 1 with ozone in CH2Cl2 without reduction of the peroxide products produced tris-nor-lactol 2 (Scheme 1). Characteristic resonances of C-20 and C-24 were observed in the 13C NMR spectrum at 8 87.6 and 99.7 ppm. A broad resonance for the C-24 OH proton at 8 4.51 ppm was characteristic of the PMR spectrum of 2. Ozonolysis of 1 in MeOH formed methoxylactol 3 in 75% yield as a 1:1 mixture of racemates at the C-24 position. The structure of 3 was inferred from the doubled resonances of C-24 of equal intensity at 8 103.1 ppm and of the OCH3 protons at 8 3.33 and 3.34 ppm.

1
We also used m-chloroperbenzoic acid (m-CPBA), dimethyldioxirane, and OsO4 as oxidants of 1. Both m-CPBA and dimethyldioxirane epoxidized the C-24(25) double bond to form a mixture of stereoisomeric epoxides 6 (ratio of α:β isomers 0.6:0.4 and 0.7:0.3, respectively) according to PMR spectra. Oxidation of 1 by OsO4 in aqueous THF produced 24,25-diol 5.
Reduction of 1 by NaBH4 in MeOH occurred with formation of 3β-hydroxydipterocarpol (4). The action of NaBH4 on 24,25-epoxide 6 produced a mixture of the oxirane ring-opening product 20,25-diol 7 and 3β-hydroxy-24,25-epoxide 8. These were separated by column chromatography. The spectrum of 8 showed resonances for the epoxy group and for the 3-hydroxy group at δ 78.9 ppm. The spectrum of 7 contained resonances for C-3 and C-24 at δ 78.94 and 75.48 ppm. Acylation of 8 by Ac2O in Py gave acetate 9 (Scheme 1).
Reaction of 1 and its 24,25-epoxide 6 with hydroxylamine hydrochloride synthesized ketoximes 10 and 11 as two isomers at the C-24(25) position according to NMR spectra (Scheme 2). Reaction of 1 with phenylhydrazine in AcOH (Fisher reaction) gave 2,3-indole 12 in 86% yield. Resonances for C-2 and C-3 at δ 140.9 and 110.3 ppm and for the aromatic substituent at δ 107.0–136.2 ppm indicated that 12 had formed. The PMR spectrum showed resonances for the aromatic protons at δ 7.02–7.81 ppm.

2
The antitumor activity (cytotoxicity) of dipterocarpol derivatives 1–4, 6, 7, and 12 was studied in vitro in 60 cell lines of nine different human tumors (lung, colon, CNS, ovary, kidney, prostate, brain, leukemia, melanoma) using the method described by the National Cancer Institute of the USA (NCI) [12–15]. Compounds were placed in cell culture medium at concentration 10−5 M for 48 h for preliminary testing. Then, growth of treated cells was compared with that of untreated control cells. Table 1 presents the results in percent growth of treated cells compared with control cells (negative values correspond to cell death). According to the criterion adopted by the NCI, compounds are considered active if they inhibit cell growth to 32% of the control or cause their death. The investigated dipterocarpol derivatives did not exhibit antitumor activity in vitro against the studied cells.
The activity of compounds 1, 5–9, and 11 were tested at the Institute of Chemistry, Vietnamese Academy of Science and Technology (IC VAST), for human epidermoid cancer KB tumor cells. The data are presented below:
Compound | IC 50 , μg/mL | Compound | IC 50 , μg/mL |
|---|---|---|---|
1 | 14.37 | 8 | 10.70 |
5 | > 128 | 9 | > 128 |
6 | > 128 | 11 | > 128 |
7 | > 128 | Ellipticine | 0.51. |
It was found for 5–7, 9, and 11 that IC50 was >128 μg/mL, i.e., substituents such as indole and oxime in ring A and lactol, epoxide, and 24,25-diol in the side chain decreased the cytotoxicity of 1. The IC50 values for 1 and its derivative 8 were 14.37 and 10.07 μg/mL. According to the selected activity criterion (IC50 < 4 μg/mL [16]), these compounds exhibited weak cytotoxicity. Introducing 3β-hydroxy and C-24,25-epoxide groups (8) into the structure of 1 enhanced insignificantly the cytotoxicity. The simultaneous presence of three hydroxyls in the C-3, C-20, and C-25 positions (7) did not affect the activity although literature data [10] indicated that several such groups were important for enhancing the activity.
Experimental
PMR and 13C NMR spectra were recorded in CDCl3 on a Bruker Avance III pulsed spectrometer (500.13 and 125.47 MHz, respectively) using a 5-mm probe with Z-gradient PABBO at constant sample temperature 298 K. Chemical shifts in NMR spectra are given in ppm relative to SiMe4 internal standard. Mass spectra were obtained on an Agilent 6310 instrument. Melting points were determined on a Boetius microstage. Specific rotation angles were taken on a Perkin-Elmer 241 MC polarimeter. Elemental analyses were performed on an EuruEA-3000 CHNS-analyzer using acetanilide as a primary standard. We used an Ozon-4K ozonator (Russia). TLC was carried out on Sorbfil plates (ZAO Sorbpolimer, Russia) using CHCl3:EtOAc (20:1). Compounds were detected by H2SO4 solution (10%) with subsequent heating at 100–120°C for 2–3 min. Compound 1 was isolated as before [17].
24( R , S )-Hydroxy-25,26,27- tris - nor -20( S )-dammar-3-one (2). Ozone (1 eq.) was passed through a solution of 1 (1 mmol, 0.44 g) in CH2Cl2 (50 mL) at −40°C until the starting material disappeared (TLC monitoring). The solvent was evaporated. The product was chromatographed over a column of Al2O3 with elution successively by C6H6 and CHCl3. Yield 0.33 g (75%) as a 1:1 mixture of two racemates (according to PMR spectra), mp 208–210°C, \( [\upalpha ]_{\mathrm{D}}^{20 }+{106^{\circ }} \) (c 0.25, CHCl3). C27H43O3. PMR spectrum (δ, ppm): 0.83 (3H × 0.5, s, CH3), 0.84 (3H × 0.5, s, CH3), 0.91 (3H, s, CH3), 0.96 (3H × 0.5, s, CH3), 0.98 (3H × 0.5, s, CH3), 0.97 (3H, s, CH3), 1.04 (3H, s, CH3), 1.08 (3H, c, CH3), 1.15–1.65 (13H, m, CH, CH2), 1.67–2.11 (7H, m, CH, CH2), 2.35–2.59 (3H, m, CH, CH2), 3.32 (1H × 0.5, m, OH), 3.68 (1H × 0.5, m, OH), 4.96 (1H, br.s, H-23). 13C NMR spectrum (δ, ppm): 15.2, 16.2, 19.6, 22.1, 24.3, 25.4, 26.7, 27.1, 28.8, 31.1, 31.7, 32.4, 32.8, 33.8, 34.1, 34.6, 36.8, 40.3, 43.5, 47.4, 49.7, 50.0, 55.3, 62.2 (0.5 × C), 61.9 (0.5 × C), 87.9 (0.5 × C-20), 87.6 (C-20),103.6 (0.5 × C-24), 103.1 (0.5 × C-24), 217.9 (C-3).
24( R , S )-Methoxy-25,26,27- tris - nor -20( S )-dammar-3-one (3). Ozone (2 eq.) was passed through a solution of 1 (1 mmol, 0.44 g) in MeOH (50 mL) at −40°C until the starting material disappeared (TLC monitoring). The solvent was evaporated. The product was chromatographed over a column of Al2O3 with elution successively by C6H6 and CHCl3. Yield 0.30 g (70%) as a 1:1 mixture of two racemates (according to PMR spectra), mp 142–145°C, \( [\upalpha ]_{\mathrm{D}}^{20 }+{36^{\circ }} \) (c 0.25, CHCl3). C28H46O3. PMR spectrum (δ, ppm, J/Hz): 0.90 (3H, s, CH3), 0.94 (3H, c, CH3), 0.98 (3H, s, CH3), 1.02 (3H, s, CH3), 1.07 (3H, s, CH3), 1.36 (3H, s, CH3), 1.11–1.64 (13H, m, CH, CH2), 1.70–2.19 (8H, m, CH, CH2), 2.46–2.75 (3H, m, CH, CH2), 3.33 and 3.34 (3H, s, OCH3, ratio 1:1), 4.93 (1H, t, J = 4.7, H-24). 13C NMR spectrum (δ, ppm ): 15.2, 16.0, 16.1, 19.6, 21.0, 21.9, 25.0, 25.5, 26.7, 26.8, 29.1, 31.1, 31.2, 34.0, 34.5, 36.8, 39.8, 40.3, 41.0, 43.3, 47.4, 49.3, 50.0, 50.1, 55.3, 88.1 and 87.9 (C-20), 104.7 and 105.2 (C-24), 217.9 (C-3).
20( S )-Hydroxydammar-24-en-3 β -ol (4). A solution of 1 (1 mmol, 0.44 g) in EtOH (30 mL) was treated with NaBH4 (2.5 mmol, 0.1 g), refluxed for 4 h, and poured into HCl solution (20 mL, 5%). The precipitate was filtered off, washed with H2O, dried, and purified by column chromatography over Al2O3 using CHCl3 eluent. Yield 0.42 g (96%), mp 134–135°C, \( [\upalpha ]_{\mathrm{D}}^{20 }+{20^{\circ }} \) (c 1.3, CHCl3) (lit. [18] mp 133–135°C). C30H52O2. PMR spectrum (δ, ppm , J/Hz): 0.69 (3H, s, CH3), 0.76 (3H, s, CH3), 0.79 (3H, s, CH3), 0.88 (3H, s, CH3), 0.89 (3H, s, CH3), 1.15 (3H, s, CH3), 1.10–1.29 (7H, m, CH, CH2), 1.31–1.49 (14H, m, CH, CH2), 1.63–1.77 (7H, m, CH, CH2), 1.91–2.30 (3H, m, CH, CH2), 3.12 (1H, dt, J1 = 5.3, J2 = 5.4, J3 = 10.7, CH), 4.21 (1H, br.s, OH), 5.12 (1H, t, J = 6.9, CH). 13C NMR spectrum (δ, ppm ): 15.4, 15.5, 16.2, 16.5, 17.7, 18.3, 21.6, 22.6, 24.8, 25.4, 25.7, 27.4, 27.6, 28.0, 31.2, 35.3, 37.1, 39.0, 39.1, 40.4, 40.5, 42.3, 49.9, 50.3, 50.7, 55.9, 75.34 (C-20), 78.94 (C-3), 124.68 (C-24), 131.63 (C-25).
20( S )-Hydroxydammar-24,25-diol-3-one (5). A solution of 1 (1 mmol, 0.44 g) in THF:H2O (10:1) was treated with N-methylmorpholine-N-oxide (NMO, 1.5 mmol) and OsO4 (0.02 mol), stirred at room temperature for 4 h, and treated with NaHSO3 solution (3 mL, 20%). The layers were separated. The aqueous layer was extracted with EtOAc (4 × 4 mL). The combined organic layers were washed until neutral and dried over Na2SO4. The solvent was evaporated in vacuo. The solid was purified by column chromatography over SiO2 using CH2Cl2:MeOH (100:1) eluent. Yield 0.34 g (78%), mp 139°C, \( [\upalpha ]_{\mathrm{D}}^{20 }-{47^{\circ }} \) (c 1.0, CHCl3). C30H52O4. PMR spectrum (δ, ppm , J/Hz): 0.88 (3H, s, CH3), 0.94 (3H, s, CH3), 1.00 (3H, d, J = 0.04, CH3), 1.04 (3H, s, CH3), 1.08 (3H, s, CH3), 1.16 (3H, s, CH3), 1.18 (3H, s, CH3), 1.23 (3H, d, J = 0.01, CH3), 1.24–1.67 (14H, m, CH, CH2), 1.68–1.95 (6H, m, CH, CH2), 2.39–2.51 (4H, m, CH), 3.40 (1H, dd, J1 = 10.4, J2 = 2.7, H-24), 4.23 (3H, br.s, 3-OH). 13C NMR spectrum (δ, ppm): 15.2, 16.0, 16.4, 16.5, 19.7, 21.0, 22.0, 24.9, 25.1, 25.4, 25.4, 26.7, 27.5, 31.1, 34.1, 34.6, 36.9, 37.0, 39.9, 40.3, 42.5, 42.6, 47.4, 50.2, 50.3, 55.4, 79.4 (C-24), 73.8 (C-25), 75.5 (C-20), 218.0 (C-3). ESI-MS, m/z: 442.1 [M (476) – H2O – OH + 1]+ (100%).
24,25( R , S )-24,25-Epoxy-20( S )-hydroxydammar-3-one (6). a) A solution of 1 (1 mmol, 0.44 g) in Me2CO (10 mL) was stirred continuously, treated in small portions with a solution of dimethyldioxirane (1.1 eq.) in Me2CO, and stirred for 3 h until the starting material disappeared completely (TLC monitoring). The solvent was evaporated in vacuo. The product was chromatographed over a column of Al2O3 with elution successively by C6H6 and CHCl3. Yield 0.36 g (83%), mp 141–143°C. PMR spectrum (δ, ppm , J/Hz): 0.93 (3H × 0.8, s, CH3), 0.94 (3H × 0.2, s, CH3), 0.99 (3H × 0.8, s, CH3), 1.01 (3H × 0.2, s, CH3), 1.03 (3H × 0.8, s, CH3), 1.04 (3H × 0.2, s, CH3), 1.08 (3H × 0.8, s, CH3), 1.09 (3H × 0.2, s, CH3), 1.12 (3H × 0.8, s, CH3), 1.13 (3H × 0.2, s, CH3), 1.14 (3H × 0.8, s, CH3), 1.15 (3H × 0.2, s, CH3), 1.19 (3H × 0.8, s, CH3), 1.21 (3H × 0.2, s, CH3), 1.25–1.70 (8H, m, CH, CH2),1.72 (3H, s, CH3), 1.73–1.95 (14H, m, CH, CH2), 2.39–2.53 (6H, m, CH, CH2), 3.75 (1H X 0.8, t, J = 7.2, H-24), 3.75 (1H × 0.2, dd, J1 = 5.5, J2 = 9.4, H-24). 13C NMR spectrum (δ, ppm): 15.1 (0.8 × C), 15.2 (0.2 × C), 16.0 (0.8 × C), 16.1 (0.2 × C), 16.3, 19.7, 21.0, 22.1 (0.8 × C), 22.3 (0.2 × C), 23.6, 24.1 (0.2 × C), 24.3 (0.8 × C), 25.7 (0.8 × C), 25.8 (0.2 × C), 26.1 (0.8 × C), 26.4 (0.2 × C), 26.7 (0.8 × C), 26.8 (0.2 × C), 27.0 (0.8 × C), 27.2 (0.2 × C), 27.4, 27.8, 31.4, 34.1, 34.6 (0.8 × C), 34.8 (0.2 × C), 35.7, 36.8, 39.9 (0.8 × C), 40.3 (0.2 × C), 43.0 (0.2 × C), 43.1 (0.8 × C), 47.4, 49.5, 49.8 (0.2 × C), 49.9 (0.8 × C), 50.0, 50.1, 55.3, 70.3, 71.4 (C-20), 83.3 (0.8 × C), 86.3 (0.2 × C), 86.4 (0.8 × C), 86.5 (0.2 × C), 217.9 (C-3).
b) A solution of 1 (1 mmol, 0.44 g) in anhydrous CHCl3 (20 mL) was treated with m-CPBA (2.6 mmol, 0.40 g), stirred in the dark for 1 d, neutralized with KI solution (10%, 2 × 20 mL) and H2O (2 × 30 mL), and dried over MgSO4. The solvent was evaporated in vacuo. The product was chromatographed over a column of Al2O3 with elution successively by C6H6 and CHCl3. Yield 0.40 g (87%), mp 142–144°C. C30H50O3. PMR spectrum (δ, ppm , J/Hz): 0.93 (3H × 0.6, s, CH3), 0.94 (3H × 0.4, s, CH3), 0.99 (3H × 0.6, s, CH3), 1.01 (3H × 0.4, s, CH3), 1.03 (3H × 0.6, s, CH3), 1.04 (3H × 0.4, s, CH3), 1.08 (3H × 0.6, s, CH3), 1.09 (3H × 0.4, s, CH3), 1.12 (3H × 0.6, s, CH3), 1.13 (3H × 0.4, s, CH3), 1.14 (3H × 0.6, s, CH3), 1.15 (3H × 0.4, s, CH3), 1.19 (3H × 0.6, s, CH3), 1.21 (3H × 0.4, s, CH3), 1.25–1.70 (8H, m, CH, CH2),1.72 (3H, s, CH3), 1.73–1.95 (14H, m, CH, CH2), 2.39–2.53 (6H, m, CH, CH2), 3.75 (1H × 0.6, t, J1 = 7.2, H-24), 3.75 (1H × 0.4, dd, J1 = 5.5, J2 = 9.4, H-24). 13C NMR spectrum (δ, ppm): 15.1 (0.6 × C), 15.2 (0.4 × C), 16.0 (0.6 × C), 16.1 (0.4 × C), 16.4, 19.7, 21.0, 22.1 (0.6 × C), 22.3 (0.4 × C), 23.4 (0.6 × C), 23.6 (0.4 × C), 24.1 (0.4 × C), 24.3 (0.6 × C), 25.7 (0.6 × C), 25.8 (0.4 × C), 26.1 (0.6 × C), 26.4 (0.4 × C), 26.7 (0.6 × C), 26.8 (0.4 × C), 27.0 (0.6 × C), 27.2 (0.4 × C), 27.5, 27.8, 31.4, 34.1, 34.6 (0.6 × C), 34.8 (0.4 × C), 35.7, 36.8, 39.9 (0.6 × C), 40.3 (0.4 × C), 43.0 (0.4 × C), 43.1 (0.6 × C), 47.4, 49.5, 49.8 (0.4 × C), 49.9 (0.6 × C), 50.0, 50.1, 55.3, 70.3, 71.4 (C-20), 83.3 (0.6 × C), 86.3 (0.4 × C), 86.4 (0.6 × C), 86.5 (0.4 × C), 218.1 (0.6 × C-3), 218.2 (0.4 × C).
Method for Synthesizing 7 and 8. A solution of 6 (1 mmol, 0.46 g) in anhydrous CH2Cl2 was stirred, cooled to 0–5°C, treated in portions with NaBH4 (2 mmol, 0.07 g), and stirred for 20 min at 5°C. The excess of NaBH4 was neutralized with HCl solution (1 M). The organic layer was separated and dried over Na2SO4. The solvent was evaporated in vacuo. The product was purified by column chromatography over SiO2 using hexane:EtOAc (5:1) eluent. Total yield of 7 and 8, 0.42 g (92%).
20( S )-Hydroxydammar-3 β ,25-diol (7). Yield 0.27 g (60%), mp 123–125°C, \( [\upalpha ]_{\mathrm{D}}^{20 }-{70^{\circ }} \) (c 1.0, CHCl3). C30H54O3. PMR spectrum (δ, ppm , J/Hz): 0.71–0.76 (2H, m, CH), 0.77 (3H, s, CH3), 0.84 (3H, s, CH3), 0.89 (3H, s, CH3), 0.96 (3H, s, CH3), 0.97 (3H, s, CH3), 1.14 (3H, s, CH3), 1.22 (3H, s, CH3), 1.25–1.37 (10H, m, CH, CH2), 1.39–1.82 (17H, m, CH, CH2), 2.05 (3H, br.s, OH), 3.19 (1H, dd, J1 = 11.2, J2 = 4.9, H-3). 13C NMR spectrum (δ, ppm ): 15.4, 15.5, 16.2, 16.5, 18.3, 18.5, 21.6, 24.9, 25.5, 27.4, 27.5, 28.0, 29.3, 29.4, 31.2, 35.2, 37.1, 38.9, 39.0, 40.4, 41.0, 42.3, 44.5, 49.8, 50.3, 50.6, 55.9, 71.1 (C-20), 75.5 (C-25), 78.9 (C-3). ESI-MS, m/z 445.6 [M (462) – H2O + 1]+ (~10%), 427.8 [M (462) –2H2O + 1]+ (100%).
20( S )-Hydroxydammar-24,25-epoxy-3 β -ol (8). Yield 0.14 g (32%) as a 3:1 mixture of two isomers, mp 158– 161°C, \( [\upalpha ]_{\mathrm{D}}^{20 }-{60^{\circ }} \) (c 1.0, CHCl3). C30H52O3. PMR spectrum (δ, ppm, J/Hz): 0.70–0.75 (2H, m, CH), 0.77 (3H, s, CH3), 0.84 (3H × 0.7, s, CH3), 0.85 (3H × 0.3, s, CH3), 0.87 (3H, s, CH3), 0.95 (3H, s, CH3), 0.97 (3H, s, CH3), 1.12 (3H × 0.7, s, CH3), 1.13 (3H × 0.3, s, CH3), 1.21 (3H, s, CH3), 1.73–1.69 (9H, m, CH, CH2), 1.69–1.91 (15H, m, CH, CH2), 2.12 (1H, br.s, CH), 3.19 (1H, dd, J1 = 5.2, J3 = 10.3, H-3), 3.39 (2H, br.s, OH), 3.65 (1H × 0.3, dd, J1 = 5.4, J2 = 9.1, H-24), 3.73 (1H × 0.7, t, J1 = 7.2, H-24). 13C NMR spectrum (δ, ppm ): 15.3, 15.4, 16.2, 16.5, 18.3, 21.5 (0.7 × C), 21.8 (0.3 × C), 23.5, 24.3, 25.7, 26.1, 27.7, 27.4, 27.3, 28.0, 31.4, 35.3, 35.7, 37.1, 38.9, 39.0, 40.3, 42.8 (0.3 × C), 42.9 (0.7 × C), 49.5, 49.8 (0.3 × C), 50.0 (0.7 × C), 50.7 (0.7 × C), 50.8 (0.3 × C), 55.8, 70.3 (0.3 × C-20), 71.43 (0.7 × C-20), 78.9 (C-3), 86.5 (0.3 × C-24), 86.4 (0.7 × C-24), 86.3 (0.3 × C-25), 83.3 (0.7 × C-25). ESI-MS, m/z 443.4 [M (460.4) – H2O + 1]+ (100%), 425.6 [M (460) –2H2O + 1]+ (100%).
20( S )-Hydroxydammar-24,25-epoxy-3 β -acetate (9). A mixture of 7 (1 mmol, 0.46 g) and anhydrous Py (2 mL) was treated with dimethylaminopyridine (0.64 g) and Ac2O (2 mL), stirred at room temperature for 2 h, and poured into H2O (20 mL). The precipitate was filtered off, washed until neutral, and purified by column chromatography over SiO2. Yield 0.43 g (94%) as a 3:1 mixture of two isomers, mp 110–111°C, \( [\upalpha ]_{\mathrm{D}}^{20 }-{9^{\circ }} \) (c 1.0, CHCl3). C32H54O4. 0.82–0.84 (2H, m, CH), 0.85 (3H × 0.7, s, CH3), 0.86 (3H × 0.3, s, CH3), 0.87 (3H × 0.7, s, CH3), 0.88 (3H × 0.3, s, CH3), 0.95 (3H, s, CH3), 0.97 (3H, s, CH3), 1.11 (3H × 0.7, s, CH3), 1.12 (3H × 0.3, s, CH3), 1.13 (3H × 0.7, s, CH3), 1.14 (3H × 0.3, s, CH3), 1.18 (3H × 0.7, s, CH3), 1.20 (3H × 0.3, s, CH3), 2.01 (3H, s, OCOCH 3), 1.24–1.38 (10H, m, CH, CH2), 1.41–1.94 (18H, m, CH, CH2), 3.72 (1H × 0.3, t, J = 7.2, H-24), 3.64 (1H × 0.7, dd, J1 = 9.9, J2 = 5.3, H-24), 4.48 (1H, m, H-3). 13C NMR spectrum (δ, ppm ): 15.4 (0.3 × C), 15.5 (0.7 × C), 16.0, 16.3 (0.7 × C), 16.4 (0.3 × C), 18.3, 21.3 (0.7 × C), 21.5 (0.3 × C), 21.8, 23.5, 23.7, 24.1, 24.3, 25.7, 25.8, 26.1, 26.4 (0.3 × C), 26.9 (0.7 × C), 27.3 (0.7 × C), 27.4 (0.3 × C), 28.0, 31.4, 35.3 (0.7 × C), 35.7 (0.3 × C), 37.1, 38.9, 39.0, 40.4, 42.8 (0.3 × C), 42.9 (0.7 × C), 49.6, 49.8 (0.3 × C), 50.0 (0.7 × C), 50.8 (0.3 × C), 50.7 (0.7 × C), 55.9, 70.3 (0.3 × C-20), 71.4 (0.7 × C-20), 80.9 (C-3), 86.5 (0.3 × C-24), 86.4 (0.7 × C-24), 86.3 (0.3 × C-25), 83.3 (0.7 × C-25), 170.9 (C=O). ESI-MS, m/z: 442.6 [M (502) – OH + 1]+ (~10%), 425.5 [M – OH – H2O + 1]+ (100%).
Method for Synthesizing 10 and 11. A solution of 1 (1 mmol, 0.44 g) or 6 (1 mmol, 0.46 g) in Py:MeOH (1:1, 20 mL) was treated with NH2OH-HCl (2.6 mmol, 1 g), refluxed for 6 h, and poured into HCl solution (20 mL, 5%). The precipitate was filtered off, washed with H2O, dried in air, and purified by column chromatography over SiO2 using CHCl2 eluent.
20( S )-Hydroxydammar-3-oxime-24-ene (10). Yield 0.37 g (85%), mp 187–188°C, R f 0.65, \( [\upalpha ]_{\mathrm{D}}^{20 }+{14^{\circ }} \) (c 1.8, CHCl3). C30H51NO2. PMR spectrum (δ, ppm , J/Hz): 0.89 (3H, s, CH3), 0.95 (3H, s, CH3), 1.01 (3H, s, CH3), 1.09 (3H, s, CH3), 1.15 (3H, s, CH3), 1.21–1.60 (14H, m, CH, CH2), 1.64 (3H, s, CH3), 1.69 (3H, s, CH3), 1.71–1.89 (11H, m, CH, CH2), 2.01–2.12 (2H, m, CH, CH2), 2.20–2.39 (1H, m, CH), 5.13 (1H, t, J = 7.1, H-24), 7.69 (1H, br.s, OH). 13C NMR spectrum (δ, ppm ): 15.3, 15.84, 16.3, 17.2, 17.6, 19.0, 21.7, 22.5, 22.8, 24.7, 25.4, 25.7, 27.3, 27.5, 31.1, 34.8, 37.1, 39.0, 40.4, 40.5, 42.3, 42.6, 49.7, 50.2 (2C), 56.0, 75.3 (C-20), 124.7 (C-24), 131.5 (C-25), 167.0 (C-3).
20( S )-Hydroxydammar-24,25-epoxy-3-oxime (11). Yield 0.38 g (82%) as a 3:1 mixture of two isomers, mp 144– 146°C, \( [\upalpha ]_{\mathrm{D}}^{20 }-{42^{\circ }} \) (c 1.0, CHCl3). C30H51NO3. PMR spectrum (δ, ppm , J/Hz): 0.86 (3H, s, CH3), 0.93 (3H, s, CH3), 0.95 (3H, s, CH3), 0.96 (3H, s, CH3), 0.98 (3H, s, CH3), 1.00 (3H, s, CH3), 1.07 (3H, s, CH3), 1.08 (3H, s, CH3), 1.11 (3H, s, CH3), 1.12 (3H, s, CH3), 1.13 (3H, s, CH3), 1.15 (3H, s, CH3), 1.16 (3H, s, CH3), 1.19 (3H, s, CH3), 1.21 (3H, s, CH3), 1.26 (3H, s, CH3), 2.42 (2H, m, Hb-2, Hb-2 isomer), 2.97 (2H, m, Ha-2, Ha-2 isomer), 3.65 (1H, dd, J1 = 9.9, J2 = 5.3, H-24), 3.73 (1H, t, J = 7.2, H-24). 13C NMR spectrum (δ, ppm): 15.8 (0.7 × C), 15.9 (0.3 × C), 16.3 (0.7 × C), 16.4 (0.3 × C), 17.4, 19.1, 21.9 (0.7 × C-24), 22.1 (0.3 × C-24), 22.9, 23.5, 24.1, 24.3, 25.7 (0.7 × C-24), 25.8 (0.3 × C-24), 26.2, 26.4, 27.0 (0.7 × C-24), 27.1 (0.3 × C-24), 27.4 (0.7 × C), 27.5 (0.3 × C), 27.5, 27.8, 29.7, 31.4, 34.9, 35.8, 37.2, 39.0, 40.4 (0.7 × C), 40.5 (0.3 × C), 42.9 (0.7 × C-24), 43.0 (0.3 × C-24), 49.6 (0.7 × C-24), 49.8 (0.3 × C-24), 50.1, 50.4 (0.7 × C-24), 50.5 (0.3 × C-24), 56.0, 70.3 (0.3 × C-20), 71.5 (0.7 × C-20), 83.4 (0.7 × C-24), 86.4 (0.3 × C-24), 86.5 (0.7 × C-25), 86.6 (0.3 × C-25), 168.0 (C-3). ESI-MS, m/z 474.3 [M + 1]+ (100%), 457.2 [M – H2O + 1]+ (~5%), 439.3 [M – 2H2O + 1]+ (~10%).
2,3-Indolo-20( S )-hydroxydammar-24-ene (12). Compound 1 (1 mmol, 0.43 g) in HOAc (20 mL) was treated with phenylhydrazine (3.5 mmol, 0.35 mL), refluxed for 15 h, and poured into stirring cold H2O. The precipitate was filtered off, washed with H2O, dried in air, and purified by column chromatography over Al2O3 using C6H6 eluent. Yield 0.36 g (86%), mp 86–88°C, \( [\upalpha ]_{\mathrm{D}}^{20 }+{76^{\circ }} \) (c 0.6, CHCl3). C36H53NO. PMR spectrum (δ, ppm , J/Hz): 0.90 (3H, s, CH3), 0.91 (3H, s, CH3), 1.05 (3H, s, CH3), 1.19 (3H, s, CH3), 1.29 (3H, s, CH3), 1.32–1.60 (14H, m, CH, CH2), 1.65 (3H, s, CH3), 1.71 (3H, s, CH3), 1.75–2.81 (12H, m, CH, CH2), 5.15 (1H, t, J = 7.1, H-24), 7.01–7.73 (4H, m, Harom.), 8.02 (1H, br.s, NH). 13C NMR spectrum.(δ, ppm ): 15.3, 15.9, 16.4, 17.7, 19.3, 22.0, 23.2, 25.7, 27.0, 27.4, 27.7, 30.9, 31.7, 33.7, 34.2, 34.8, 37.6, 38.3, 40.5, 40.9, 44.5, 49.3, 49.8, 50.2, 53.8, 76.6 (C-20), 107.1 (Carom.), 110.3 (C-2), 118.0 (Carom.), 121.0 (Carom.), 123.9 (C-24), 127.3 (Carom.), 130.0 (C-25), 136.2 (Carom.), 137.3 (Carom.), 140.9 (C-3).
The method for testing the in vitro antitumor activity of 1–4, 6, 7, and 12 on 60 cell lines of nine human tumors is available at the website www.dtp.nci.nih.gov.
The cytotoxicity of 1, 5–9, and 11 was studied at the IC VAST using epidermoid cancer KB cells and the standard MTT test [19] that is based on the ability of dehydrogenases of living cells to transform the colorless form of MTT reagent [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] into blue crystals of formazan that are soluble in DMSO.
Cell lines of epidermal cancer KB were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were grown in DMEM (Dulbecco’s modified Eagle’s medium) with added fetal calf serum (10%) and penicillin and streptomycin (1%) at 37°C, 5% atmospheric CO2, and 95% humidity. Cells were treated with the studied compounds at the required concentration after 24 h and incubated for 48 h. MTT reagent (5 mg/mL) was added at the end of the incubation period. Optical absorption was measured on a Tecan Geniou instrument at 450 nm. Results were evaluated against control samples (without added compound) and expressed as the cell survival index in percent that was calculated using the formula At/Ac × 100, where At is the optical absorption of the test sample and Ac, that of the control. The IC50 value was determined from a dose–effect curve as the lowest concentration at which growth of the treated cells was reduced by 50% compared with the control cells. A compound was considered active if IC50 < 4 μg/mL [16].
References
T. Akihisa, H. Tokuda, M. Ukiya, T. Suzuki, F. Enjo, K. Koike, T. Nikado, and H. Nishino, Chem. Pharm. Bull., 52, 153 (2004).
F. Inada, M. Somekawa, and H. Murata, Chem. Pharm. Bull., 41, 617 (1993).
G. V. Platonov, A. D. Zorina, M. A. Gordon, N. P. Chizhov, L. V. Balykina, Yu. D. Mikhailov, D. R. Ivanen, T. K. Qui, and A. G. Shavva, Khim.-farm. Zh., 29, 42 (1995).
T. Akihisa, J. Ogihara, J. Kato, K. Yasukawa, M. Ukiya, S. Yamanouchi, and K. Oishi, Lipids, 36, 507 (2001).
D. Scholz, K. Baumann, M. Grassberger, B. Wolff-Winiski, G. Rihs, H. Walter, and J. G. Meingassner, Bioorg. Med. Chem. Lett., 14, 2983 (2004).
J.-M. Zhao, N. Li, C.-F. Wu, H.-R. Piao, and Y.-Q. Zhao, Bioorg. Med. Chem. Lett., 21, 1027 (2011).
N. H. Tung, G. Y. Song, J.-A. Kim, J.-H. Hyun, H.-K. Kang, and Y. H. Kim, Bioorg. Med. Chem. Lett., 20, 309 (2010).
J. Phongmaykin, T. Kumamoto, T. Ishikawa, E. Saifah, and R. Suttisri, Nat. Prod. Res., 25 (17), 1621 (2011).
M. Ukiya, T. Kikuchi, H. Tokuda, K. Tabata, Y. Kimura, T. Arai, Y. Ezaki, O. Oseto, T. Suzuki, and T. Akihisa, Chem. Biodiversity, 7, 1871 (2010).
D. Schmitz, J. Zapp, and R. Bernhardt, FEBS J., 279, 1663 (2012).
H. Hasegawa, J. Pharmacol. Sci., 95, 153 (2004).
M. C. Alley, D. A. Scudiero, P. A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker, and M. R. Boyd, Cancer Res., 48 (3), 589 (1988).
M. R. Grever, S. A. Schepartz, and B. A. Chabner, Semin. Oncol., 19, 622 (1992).
M. R. Boyd and K. D. Paull, Drug. Dev., 34, 91 (1995).
R. H. Shoemaker, Nat. Rev. Cancer, 6, 813 (2006).
C. C. Lee and P. Houghton, J. Ethnopharmacol., 100, 237 (2005).
L. Q. Tran and K. Q. Tran, J. Chem., 36, 8 (1998).
A. D. Zorina, L. V. Balykina, O. V. Nazarova, and A. A. Rebezov, Zh. Prikl. Khim., 79, 663 (2006).
D. Sladowski, S. J. Steer, R. H. Chothier, and M. Balls, J. Immunol. Methods, 157, 203 (1993).
Acknowledgment
The work was supported financially by grants of the National Foundation for Science and Technology Development of Vietnam (Nafosted) and the RFBR No. 10-03-90303 and No. 11-03-12144. We thank the NCI for determining the in vitro antitumor activity of 1–4, 6, 7, and 12.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 1, January– February, 2013, pp. 51–57.
Rights and permissions
About this article
Cite this article
Huong, D.T.T., Thuy, T.T.T., Hien, T.T. et al. Synthesis and Cytotoxicity of Derivatives of Dipterocarpol, a Metabolite of Dipterocarpus alatus . Chem Nat Compd 49, 58–65 (2013). https://doi.org/10.1007/s10600-013-0505-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-013-0505-4